INTRODUCTION
La granulomatose avec polyangéite (GPA) est une maladie rare avec une prévalence comprise entre 1/42000 et 1/6400 habitants et son incidence annuelle varie de 2-12 cas par million. Les séries historiques documentent une mortalité de 80% à 2 ans, en l’absence de traitement. Au niveau anatomopathologique, c’est une vascularite nécrosante des vaisseaux de petit et moyen calibres associant une inflammation de la paroi et des granulomes péri- et extravasculaires. La maladie atteint préférentiellement les voies aériennes (tant supérieures qu’inférieures) et le rein, mais elle peut s’étendre potentiellement à tous les organes. Le pronostic est souvent lié à la sévérité de l’atteinte rénale.
CAS CLINIQUE
Mme V., âgée de 37 ans, est admise aux urgences dans un tableau de céphalées importantes accompagnées de nausées et de vomissements. La patiente se plaint également d’otalgies bilatérales persistantes associées à des otorrhées purulentes, malgré plusieurs traitements antibiotiques locaux et systémiques. À ses plaintes initiales s’ajoutent une asthénie, un état subfébrile et une perte pondérale progressive (estimée à 5-6kg) sur plusieurs semaines. Ses antécédents sont sans particularité. L’examen clinique objective uniquement une hyperthermie à 38.3°C et une sensibilité de la nuque à la flexion, sans raideur et sans autre anomalie à l’examen neurologique. Biologiquement, on retiendra un important syndrome inflammatoire (CRP à 45 mg/dl (VN : < 0,5) sans hyperleucocytose associée mais avec une petite anémie (Hb à 11.5 g/dl (VN entre 12 et 16 g/dl) et une thrombocytose (660000/mm³ (VN : 150-400000). Les fonctions rénales (créatininémie à 0,8 mg/dl (VN < 0,9) et hépatiques ainsi que l’ionogramme sont dans les normes. Devant ce tableau, une ponction lombaire est réalisée mais s’avère rassurante (quelques lymphocytes (12/µl), un glyco- et une protéinorachie normales, culture stériles). Un sédiment urinaire réalisé dans le contexte fébrile met en évidence une hématurie microscopique (50 GR/champ), avec présence de globules rouges dysmorphiques et d’une protéinurie conséquente (54 mg/dl (VN 0-10)). La radiographie de thorax est normale. Le scanner cérébral visualise une oto-mastoïdite bilatérale ainsi qu’une sinusite principalement maxillaire droite, mais pas d’atteinte du parenchyme cérébral. (Fig. 1)
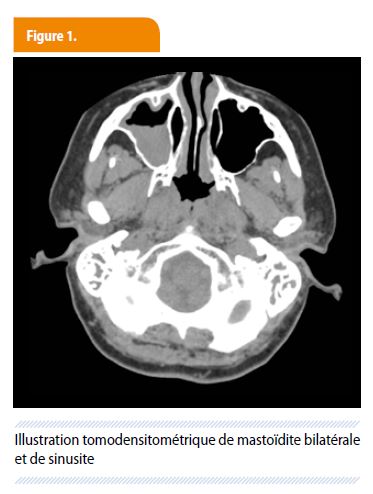
Le syndrome inflammatoire important associé aux signes cliniques de méningisme fait craindre une méningite décapitée par le traitement antibiotique antérieur et la patiente est hospitalisée dans le service de médecine interne générale pour un traitement par Rocéphine et Clamoxyl intra-veineux. Un bilan ORL sera rapidement réalisé, avec drainage de l’oreille moyenne. Face à ce tableau d’atteinte pluri-organique et l’importance du syndrome inflammatoire, une recherche de maladie systémique est lancée. La sérologique ANCA s’avère positive, avec un titre à 1/320 (VN : < 40). Les Ac anti-MPO négatifs et des Ac anti-PR3 positifs à 191 U/ml (positif > 25, fortement positif > 80 U/ml). En présence d’une atteinte ORL, des anomalies du sédiment urinaire et d’ANCA anti-PR3 positifs, le diagnostic de granulomatose avec polyangéite est posé et un traitement d’induction classique instauré : corticoïdes à hautes doses (Solumédrol 1g 1x/j pendant 3 jours, suivi par 48 mg de Médrol par jour) associé à du Cyclophosphamide (en pulse thérapie 750mg 1x/mois, soit 7,5mg/kg).
EVOLUTION
Endéans les 48 heures du début du traitement, la patiente développe une cécité droite correspondant, à une thrombose de l’artère centrale de la rétine droite. Une thrombophilie d’origine génétique ou auto-immunue a été exclue. L’hypothèse d’une thrombose favorisée par la thrombocytose dans un contexte d’inflammation chronique est retenue. Une récupération partielle de l’acuité visuelle droite est obtenue par le traitement antiagrégant plaquettaire qui sera poursuivi au long cours.
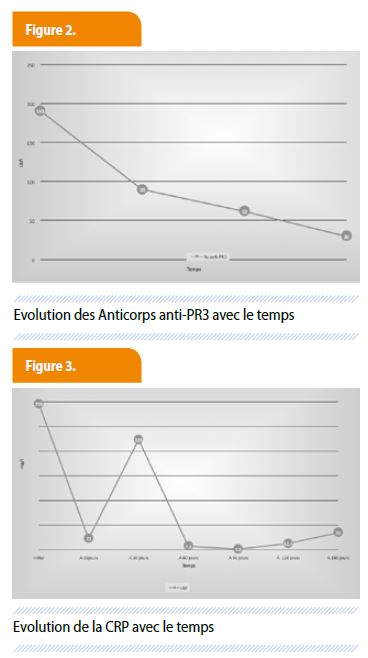
Environ trois semaines plus tard, la patiente se présentera aux urgences pour de fortes douleurs dans le flanc et l’hypochondre gauches. Le bilan tomodensitométrique mettra en évidence un infarctus splénique étendu. Après concertation pluri-disciplinaire, décision de ne pas réaliser de splénectomie, mais d’une surveillance hospitalière et mise en route d’une anticoagulation. Durant la surveillance, et malgré une évolution favorable tant clinique (disparition des plaintes ORL initiales, disparition des douleurs abdominales) que biologique (diminution du syndrome inflammatoire et du taux d’Ac anti-PR3) (Fig. 2 et 3), la patiente signale des palpitations. Si l’ECG simple s’avère sans particularité, un Holter-ECG de 24 heures montre un bloc auriculo-ventriculaire du 3ème degré avec des pauses prolongées répétées allant jusqu’à 30 secondes (Fig. 4) justifiant l’implantation d’un stimulateur cardiaque.

Au bout de 5 mois de traitement d’attaque une rémission clinico-biologique et atteinte et un traitement d’entretien instauré.
DISCUSSION
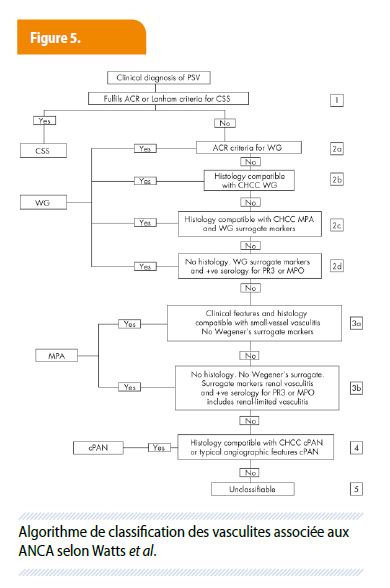
Le diagnostic de granulomatose avec polyangéite est basé sur les critères établis par l’American college of rheumatology, auxquels a été ajouté la sérologie ANCA (Fig. 5)1-2. On retiendra par ailleurs que de nouveaux critères diagnostics (incluant les ANCA PR3) en cours d’élaboration (DCVAS study)3.
Dans notre cas, l’analyse des biopsies réalisées au niveau des sinus et du liquide de drainage auriculaire s’avérera non contributif, mais l’atteinte ORL combinée à l’atteinte rénale et à la positivité de la sérologie permettra de poser le diagnostic. Une fois le diagnostic posé, un traitement à base de corticoïdes et de Cyclophosphamide (en pulse thérapie IV) a été décidé, sur bases des recommandations actuelles.4-5
Cependant, malgré la précocité du traitement et son efficacité biologique, la patient sera victime, successivement d’une thrombose de l’artère centrale de la rétine, d’un infarctus splénique et d’un bloc auriculo-ventriculaire du 3ème degré.
L’atteinte ophtalmologique survient chez 30 à 50 % des patients souffrants de granulomatose avec polyangéite et peut toucher n’importe quelle partie de l’œil, allant de la simple conjonctivite jusqu’à la sclérite nécrosante, les ulcérations cornéennes ou la diplopie, en passant par les uvéites ou une névrite optique6.
Une atteinte vasculaire ophtalmique reste rare, avec très peu de cas décrits. Une importante revue de la littérature datant de 2007, n’évoquait, à l’époque que moins de 20 cas recensés.7 Il n’existe actuellement pas de traitement spécifique pour cette pathologie, et la récupération visuelle reste très variable d’un patient à l’autre.
L’atteinte splénique dans la vasculite avec polyangéite inclus différents types d’anomalies, allant de la simple splénomégalie à l’infarctus massif. Bien que fréquemment découverte lors d’autopsies, il n’existe que peu de cas rapportés chez des patients vivants, ceci pouvant s’expliquer par la fréquence élevée d’atteintes asymptomatiques ainsi, 36 cas d’infarctus splénique ont été publiés dans la littérature et seulement 8 des 16 cas où nous disposons d’informations cliniques ont été symptomatiques8. L’infarctus splénique prédispose également à plusieurs complications secondaires telles qu’un risque infectieux accru, des abcès ou encore une rupture spontanée de la rate8. La physiopathologie de ces phénomènes thrombotiques reste actuellement peu claire, bien qu’un rôle des ANCA soit évoqué, notamment via les interactions directes sur l’endothélium et sur la réponse neutrophilique9. Par ailleurs, la corticothérapie à hautes doses participe également au risque thrombogène (notamment par induction du facteur VIII et de la fibrinolyse)10.
Les complications cardiaques sont présentes que chez environ 10% des malades atteints de granulomatose avec polyangéite, et se comportent principalement de péricardites, de vascularite des artères coronaires et de myocardites. Les troubles du rythme représentent une plus faible part des atteintes cardiaques et leur gravité est très variable, allant du simple bloc de branche asymptomatique au bloc auriculo-ventriculaire complet, qui reste très rare (seulement quelques cas décrits)11-14. De plus, l’atteinte cardiaque est reconnue comme facteur de mauvais pronostic de la maladie, notamment parce qu’elle témoigne d’une atteinte diffuse de la maladie11,15.
CONCLUSIONS
Il existe dans notre cas, une réelle discordance entre l’atteinte initiale essentiellement ORL, la bonne réponse biologique au traitement initié et l’évolution de la maladie. En effet, durant le premier mois suivant le diagnostic et sous traitement d’induction, la patiente a présenté plusieurs manifestations systémiques rares et sévères. Bien qu’elles n’aient pas entraîné d’issue fatale, ces dernières ont considérablement majoré la morbidité de cette jeune patiente.
Recommandations pratiques
Outre l’introduction d’un traitement précoce et respectant les dernières recommandations, un suivi régulier reste capital, de même qu’un bilan minutieux de chaque plainte du patient souffrant d’une granulomatose avec polyangéite afin de ne pas grever sa morbi-mortalité.
Affiliations
1. Service de néphrologie et médecine interne générale, CHU UCL Namur, Belgique
Correspondance
Dr. Nathan Scius
CHU UCL Namur - site Godinne
Médecine Interne Générale
Avenue G.Thérasse,1-B-5530 Yvoir
Références
- Leavitt RY, Fauci AS, Bloch DA, et al. “The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis”. Arthritis Rheum. 1990 33 (8): 1101–7.
ouvrir dans Pubmed - Watts R, Lane S, Hanslik T et al. Development and validation of a consensus methodology for the classification of the ANCA associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007;66:222–7.
ouvrir dans Pubmed - Luqmani R, Watts et al. Nomenclature and classification of vasculitis – update on the ACR/EULAR Diagnosis and Classification of Vasculitis Study (DCVAS). Clin Exp Immunol. 2011 May; 164(Suppl 1): 11–13.
ouvrir dans Pubmed - Yates M, Watts RA, Bajema IM, et al. EULAR/ERA -EDTA recommendations for the management of ANCA -associated vasculitis. Ann Rheum Dis. 2016; 75: 1583-94.
ouvrir dans Pubmed - De Groot K, Harper L, Jayne D et al. “Pulse Versus Daily Oral Cyclophosphamide for Induction of Remission in Antineutrophil Cytoplasmic Antibody—Associated Vasculitis, A Randomized Trial”. Ann Intern Med. 2009 May; 150(10):670-680.
ouvrir dans Pubmed - Kubaisi B, Abu Samra K, Foster CS. Granulomatosis with polyangiitis (Wegener’s disease): An updated review of ocular disease manifestations; Intractable Rare Dis Res. 2016 May; 5(2): 61–69.
ouvrir dans Pubmed - Morell-Dubois S, Quéméneur T, Bourdon F, Lambert M, Queyrel V, Launay D, Hachulla E, Labalette P, Hatron PY. « Central retinal artery occlusion in Wegener’granulomatosis ». Rev Med Interne. 2007 Jan; 28(1):33-7.
ouvrir dans Pubmed - Ghinoi A, Pipitone N, Cavazza A, Boiardi L, Salvarani C. « Wegener granulomatosis with spleen infarction: case report and review of the literature ». Semin Arthritis Rheum. 2008 Apr; 37(5):328-33.
ouvrir dans Pubmed - Kelley JM, Edberg JC, Kimberly RP. Wegener’s granulomatosis: a model of auto-antibodies in mucosal autoimmunity. Clin Immunol. 2010; 134:104–112.
ouvrir dans Pubmed - Turnbull J, Harper L. « Adverse effects of therapy for ANCA-associated vasculitis ». Best Pract Res Clin Rheumatol. 2009 Jun; 23(3):391-401.
ouvrir dans Pubmed - Sarlon G, Durant C, Grandgeorge Y, Bernit E, Veit V, Hamidou M, Schleinitz N, Harlé JR. « Cardiac involvement in Wegener’s granulomatosis: report of four cases and review of the literature ». Rev Med Interne. 2010 Feb;31(2):135-9.
ouvrir dans Pubmed - Forstot JZ, Overlie PA, Neufeld GK, Harmon CE, Forstot SL. “Cardiac complications of Wegener granulomatosis: a case report of complete heart block and review of the literature”. Semin Arthritis Rheum. 1980 Nov;10(2):148-54.
ouvrir dans Pubmed - Ghaussy NO, Du Clos TW, Ashley PA. Limited Wegener’s granulomatosis presenting with complete heart block. Scand J Rheumatol. 2004;33(2):115-8.
ouvrir dans Pubmed - Elikowski W1, Baszko A, Puszczewicz M, Stachura E. Complete heart block due to Wegener’s granulomatosis: a case report and literature review. Kardiol Pol. 2006 Jun;64(6):622-7.
ouvrir dans Pubmed - Oliveira GH, Seward JB, Tsang TS, Specks U. “Echocardiographic findings in patients with Wegener granulomatosis”. Mayo Clin Proc. 2005 Nov;80 (11):1435-40.
ouvrir dans Pubmed