INTRODUCTION
Les éosinophiles sont des globules blancs issus de la lignée granulocytaire, présents essentiellement au niveau tissulaire et dont la différenciation dépend des interleukines (IL) 3 et 5 et du GM-CSF (granulocyte-macrophage colony-stimulating factor). Ils interviennent principalement dans les réactions allergiques et les maladies parasitaires. Une éosinophilie correspond à une augmentation du taux circulant ≥ 500/µL et est classée en légère (500-1500/µL), modérée (1500-5000/µL) ou sévère (>5000/µL) (1). Nous parlons d’hyperéosinophilie à partir d’une élévation sanguine modérée (>1500/µL) retrouvée à 2 reprises à 1 mois d’intervalle ou d’une atteinte tissulaire (éosinophiles médullaires >20% et/ou infiltration tissulaire extensive et/ou dépôt tissulaire de protéines d’origine granulaire) (2). La définition du syndrome hyperéosinophilique n’est pas sujette à consensus dans le milieu scientifique actuellement. Pour cet article, nous retiendrons celle-ci : un syndrome hyperéosinophilique (HES) est défini par la présence d’une hyperéosinophilie (comme sus-décrite) associée à une atteinte organique subséquente après exclusion d’une autre étiologie pour cette dernière (3,4). Le syndrome hyperéosinophilique est rare et son incidence est estimée à 0,036/100.000 (après exclusion des causes secondaires). L’âge au moment du diagnostic se situe généralement entre 20 et 50 ans, et le ratio homme/femme est de 1,47 (1).
La toxicité tissulaire des éosinophiles est médiée par la libération de médiateurs, d’enzymes, de radicaux libres mais surtout de protéines cytotoxiques : MBP (major basic protein), ECP (eosinophil cationic protein), EDN (eosinophil derived neurotoxine) et EPX (eosinophil peroxidase). L’atteinte cardiaque aigue nécrotique est secondaire à cette dégranulation alors que le stade thrombotique semble lié à des lésions endothéliales avec exposition du facteur tissulaire (5).
CAS CLINIQUES
CAS N°1
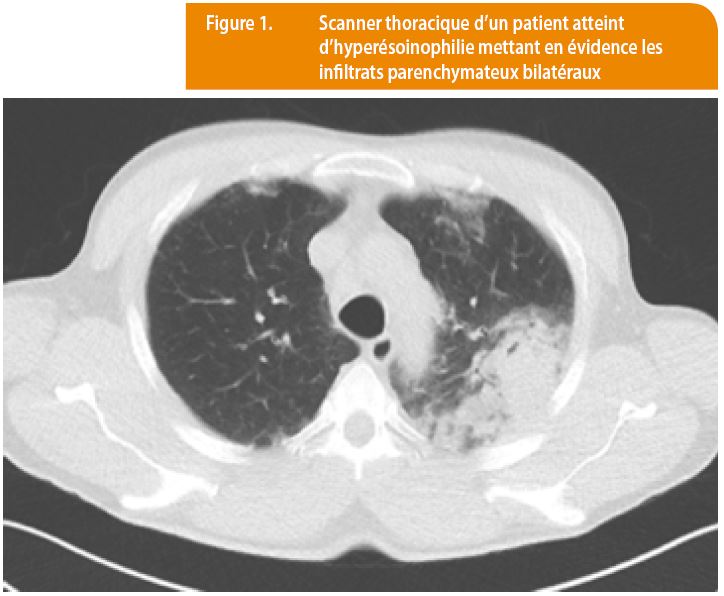
Un patient de 54 ans sans antécédent particulier se présente en consultation pour une toux sèche chronique évoluant depuis un séjour aux Seychelles 1 an auparavant, sans dyspnée ni douleur thoracique. L’état général est conservé. Il n’y a pas d’exposition professionnelle ni de tabagisme et aucune médication. L’hémogramme révèle des taux d’érythrocytes et de plaquettes dans les normes, une leucocytose à 19800/µl avec une hyper-éosinophilie à 11000/µl, le taux de lymphocytes est de 990/µl, les neutrophiles 6540/µl, les monocytes 990/µl et les basophiles 0/µl. La troponine est majorée à 0,492 ng/ml et le CEA à 570 ng/ml. Le bilan auto-immun est négatif, de même que les sérologies infectieuses, la coproculture et la culture d’expectorations. La ponction-biopsie médullaire révèle une hyperplasie de la lignée éosinophile. Le caryotype est normal et il n’y a pas de mutation FIP1L1-PDGFR en FISH. Le scanner thoracique montre un important infiltrat apical bilatéral (Figure 1) et l’IRM cardiaque une atteinte endo-myocardique. Des lésions méningées sont découvertes à l’IRM cérébrale. Le diagnostic retenu est un syndrome hyperéosinophilique primaire avec atteintes cardiaque, pulmonaire et cérébrale. Sur le plan thérapeutique, l’administration de corticoïdes a initialement donné une excellente réponse puis le développement d’un syndrome cushingoïde a nécessité un changement vers l’Hydrea et l’interféron alpha. Enfin en raison d’une intolérance à ces derniers (céphalées et troubles gastro-intestinaux invalidants), un traitement par anti-IL5, le mepolizumab (Nucala), a été débuté avec une rémission clinique et biologique soutenue de 8 mois.
CAS N°2
Un patient de 50 ans est adressé en hématologie pour bilan d’hyperéosinophiliede découverte fortuite. Il n’a aucune plainte et l’examen clinique est sans particularité. Il n’a pas d’antécédents notables à l’exception d’un tabagisme stoppé 20 ans plus tôt et d’une exposition professionnelle occasionnelle à des solvants. Le traitement habituel est composé d’antalgiques au besoin. La biologie sanguine révèle une hyperéosinophilie de 2150/mm3 et une hypergammaglobulinémie polyclonale. L’immunophénotypage lymphocytaire est normal, les sérologies infectieuses et le bilan auto-immun négatifs. La ponction de moelle retrouve une hyperplasie éosinophile. Le caryotype est normal et il n’y a pas de mutation BCR-ABL ou FIP1L1-PDGFR. Le scanner thoraco-abdomino-pelvien est rassurant. Le patient a bénéficié d’une simple surveillance régulière.
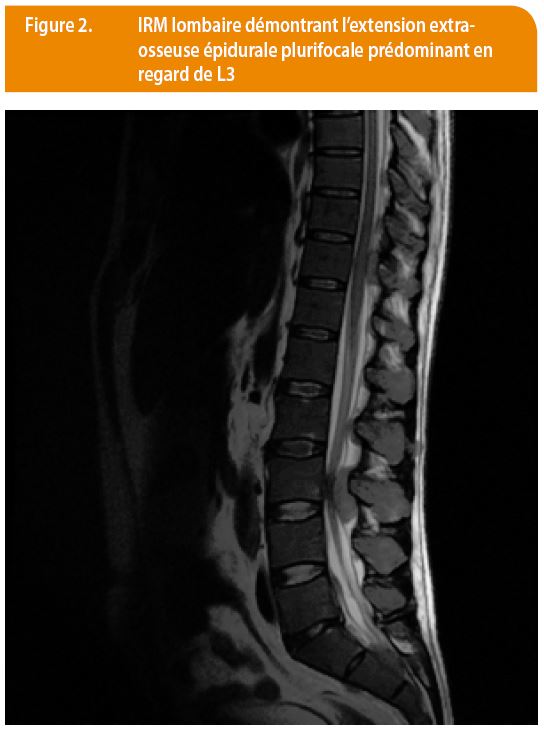
Un an plus tard est réalisé un scanner lombo-sacré suite à l’apparition de lombalgies et d’une parésie du membre inférieur droit. L’examen révèle de multiples lésions lytiques avec extension épidurale et compression médullaire (Figure 2). La biologie sanguine retrouve des précurseurs myélocytaires et une population blastique. La ponction médullaire montre une myéloblastose de 12%. Ces éléments plaident en faveur d’un syndrome myéloprolifératif de type leucémie myéloïde chronique atypique en phase accélérée. Le patient est 47,XY,+8 avec perte du chromosome 17p en cytogénétique, ce qui est de mauvais pronostic. La biologie moléculaire ne retrouve pas de mutation JAK2 ni BCR-ABL. Un chlorome est biopsié et l’analyse confirme le diagnostic de leucémie myéloïde aigue. Après une chirurgie décompressive du rachis le patient bénéficie d’une chimiothérapie puis d’une allogreffe. L’évolution s’avère défavorable et le patient décède 10 mois plus tard d’une maladie du greffon contre l’hôte et de complications infectieuses.
CAS N°3
Un patient de 46 ans se présente en hématologie pour une hyperéosinophilie isolée sans aucune plainte associée, évoluant depuis plusieurs années, supposée initialement d’origine atopique. De ses antécédents nous retenons un traumatisme crânien 10 ans auparavant ainsi qu’une rhinite allergique saisonnière. Il n’y a pas d’habitude éthylo-tabagique, pas de voyage récent ni de modification médicamenteuse. L’examen clinique est sans particularité. L’hyperéosinophilie s’élève à 3190/mm3, le typage lymphocytaire retrouve une hyperlymphocytose T CD4 et on décèle un pic monoclonal IgG lambda. Les sérologies virales et parasitaires sont négatives, de même que les RAST et les facteurs auto-immuns, et le taux d’IgE totales est normal. On observe une hyperéosinophilie marquée et une plasmocytose de 6% à la ponction-biopsie médullaire. Le caryotype est normal. La recherche de mutation FIP1L1-PDGFR en biologie moléculaire est positive. Le scanner corps entier ne montre aucune anomalie. Nous concluons à un syndrome hyperéosinophilique avec translocation FIP1L1-PDGFR (sans évidence d’atteinte d’organe) ainsi qu’à une M-GUS. La mutation étant retrouvée chez le patient, celui-ci entame un traitement ciblé par imatinib, un inhibiteur de tyrosine kinase. Le traitement est bien toléré et on assiste à une normalisation de l’hémogramme à 2 mois et une disparition de la mutation FIP1L1 à 5 mois. Le patient est toujours en rémission après 3 ans et la M-GUS reste stable.
DISCUSSION
PHYSIOPATHOLOGIE ET CLASSIFICATION
L’hyperéosinophilie peut être subdivisée selon son origine en : primaire ou clonale (prolifération néoplasique de cellules de la lignée myéloïde), secondaire ou réactive (prolifération polyclonale cytokines-médiée via IL-5, IL-3 ou GM-CSF), héréditaire (agrégation familiale après exclusion de causes primaires ou secondaires) ou enfin de signification indéterminée (après exclusion des causes primaires, secondaires ou héréditaires et en l’absence d’une lésion d’organe associée) (2).
Les étiologies secondaires comprennent par ordre de fréquence dans les pays industrialisés (1,5):
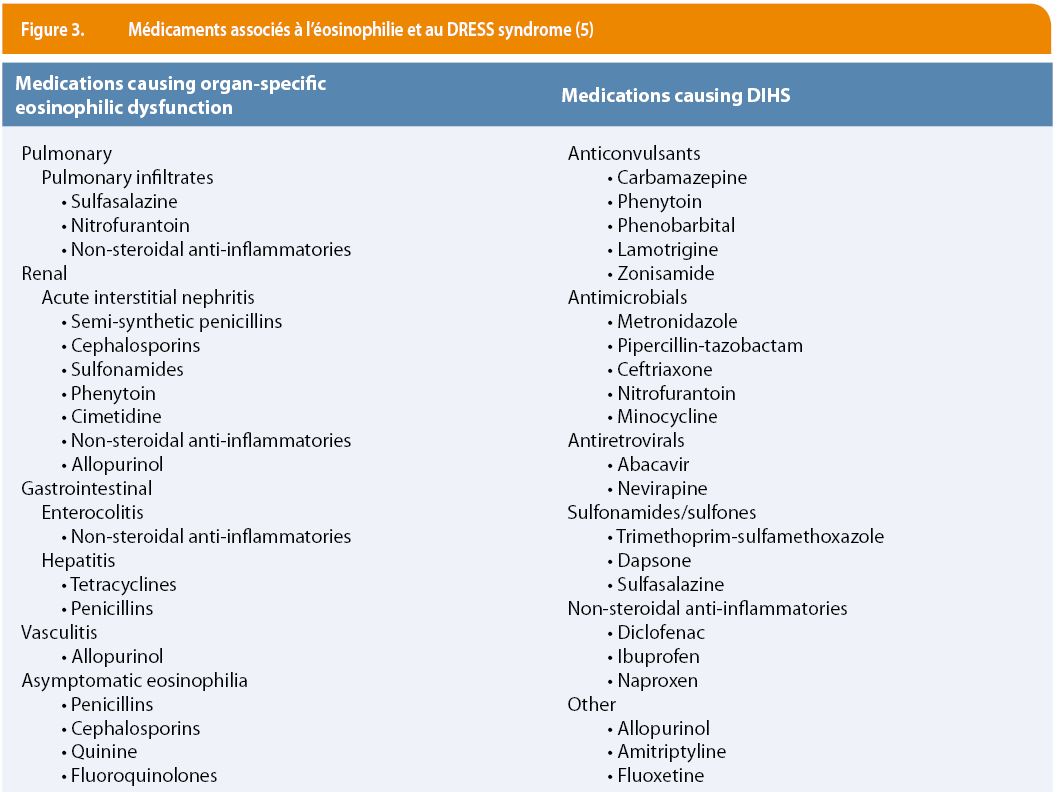
- les réactions médicamenteuses : certaines classes thérapeutiques sont plus à risque (Figure 3) mais potentiellement tout médicament peut être incriminé. Le DRESS (pour « drug rash eosinophilia and systemic symptoms ») est la forme avec atteinte d’organe-cible, et débute généralement dans les 3 mois après l’initiation du médicament responsable (5) ;
- les allergies : asthme, rhinosinusites chroniques, dermatites, aspergillose broncho-pulmonaire allergique ;
- les néoplasies hématologiques ou non hématologiques : leucémie myéloide chronique, mastocytose systémique, leucémie aiguë myéloblastique ou lymphoblastique, lymphomes B ou T mais aussi certains adénocarcinomes ;
- les infections parasitaires : essentiellement strongyloidose, toxocarose et trichinellose qui sont retrouvées dans le monde entier, alors que l’ankylostomose, la filariose ou la schistosomiase ne sont pas présentes en Europe de l’ouest ;
- les variants lymphoïdes T des syndromes hyperéosinophiliques (L-HES) : ils sont classés parmi les causes d’HES secondaires, la production d’IL-5 est liée à une population de lymphocytes anormaux (le plus souvent des lymphocytes avec un phénotype CD3-CD4+ ou CD3+CD4-CD8- mais d’autres phénotypes peuvent être identifiés) (5). On retrouve également un taux d’IgE sérique augmenté et parfois une hypergammaglobulinémie polyclonale. Les patients atteints sont à risque de développer ultérieurement une leucémie ou un lymphome T et doivent être surveillés (4) ;
- les maladies de système : surtout la granulomatose éosinophilique avec polyangéite ;
- les infections non parasitaires : HTLV-1 et 2, infection mycotique (aspergillose, coccidioïdomycose) ;
- l’insuffisance surrénalienne : en rapport avec l’hypocorticisme, qu’il soit primaire ou secondaire ;
- le syndrome lymphoprolifératif auto-immun (ALPS) ;
- la réaction du greffon contre l’hôte (GVHD) chez un patient greffé.
Parmi les étiologies primaires nous trouvons (2,5) :
- les néoplasies issues de la lignée myéloïde associées à une éosinophilie : certaines anomalies touchant spécifiquement les gènes PDGFRA, PDGFRB, FGFR1 sont recherchées (2,6). La plus fréquente consiste en la fusion des gènes FIP1L1 et PDGFRA. De cette fusion résulte une activation tyrosine kinase à la base de la prolifération et par conséquent une thérapie ciblée par inhibiteurs de tyrosine kinase pourra être proposée chez ces patients ;
- la leucémie chronique à éosinophiles : on retrouve une prolifération clonale d’éosinophiles dysplasiques avec des anomalies cytogénétiques n’entrant pas dans les autres catégories, et une population blastique (>2% dans le sang ou >5% dans la moelle) (1).
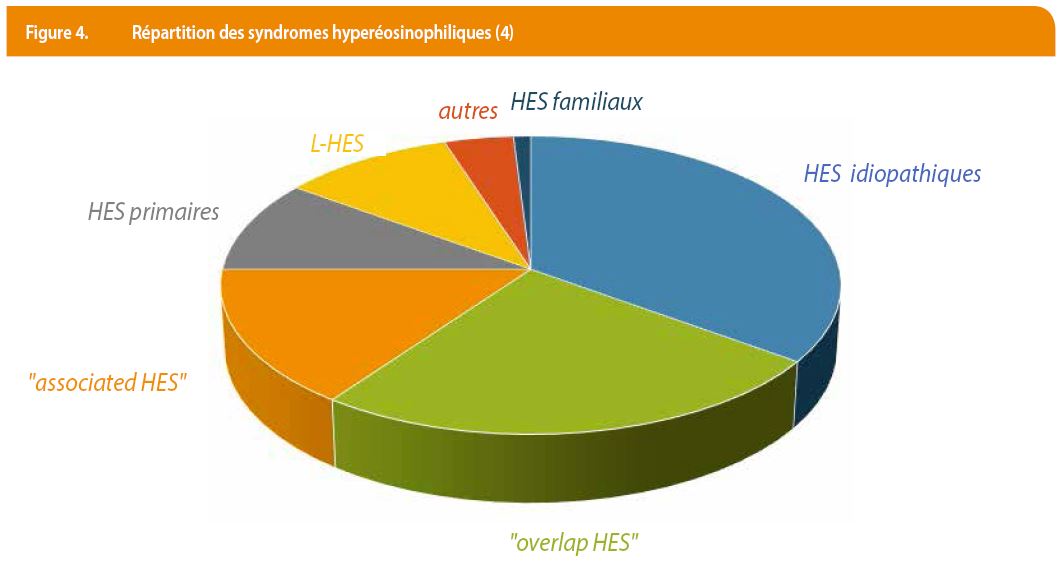
Comme défini précédemment les HES correspondent à une hyperéosinophilie associée à une atteinte organique. Ils sont le plus souvent d’étiologie primaire mais des causes secondaires sont également possibles (par exemple une réaction médicamenteuse (DRESS) ou une infection parasitaire de type syndrome de Löffler). Les HES d’origine secondaire clairement identifiée sont appelés « associated HES ». Sont également décrits les variants idiopathiques (I-HES) et les variants familiaux (de transmission autosomale dominante). Enfin l’« overlap HES » est défini par une hyperéosinophilie sanguine avec atteinte d’un seul organe-cible (4) (Figure 4). Ainsi nous obtenons comme diagnostic pour les cas cliniques : un HES primaire pour les cas n°1 et 2, et le cas n°3 correspond à une hyperéosinophilie et pas un HES en raison de l’absence d’atteinte organique.
PRÉSENTATION CLINIQUE
Par ordre de fréquence les organes atteints sont (1,5):
- la peau et les muqueuses (eczéma, urticaire, angioedème, érythrodermie, ulcères des muqueuses, prurit) – 69% ;
- le tractus respiratoire (embolie pulmonaire, adénopathies médiastinales, épanchements pleuraux, fibrose avec infiltrats parenchymateux en verre dépoli au scanner thoracique ; les principaux symptômes étant la dyspnée, la toux et les sibilances) – 44% ;
- le système digestif (gastro-entérite, colite, hépatite chronique, cholangite, se manifestant par une perte de poids, des vomissements, de la diarrhée, une dysphagie ou une perte de poids) – 38% ;
- le cœur (endocardite et myocardite nécrosantes évoluant vers la formation de thrombi intra-cavitaires puis enfin vers la fibrose avec valvulopathies, ruptures de cordages et signes de décompensation cardiaque). L’atteinte cardiaque constitue la principale cause de mortalité des HES (7) – 20% ;
- le système nerveux (AVC, neuropathies périphériques ou encéphalopathies) ;
- hypercoagulabilité et risque thrombotique (thromboses artérielles ou veineuses, nécrose des extrémités) ;
- les atteintes rhumatologiques (arthrite, syndrome de Raynaud).
Les néoplasies myéloprolifératifs ont tendance à se présenter par une atteinte cardiaque et neurologique prédominante alors que les variants lymphoïdes montrent plutôt des lésions cutanéo-muqueuses de type érythrodermie, eczéma ou urticaire (4,5).
DIAGNOSTIC
La démarche initiale vise à rechercher une étiologie secondaire et une atteinte organique (parfois asymptomatique).
Une fois l’hyperéosinophilie mise en évidence la prise en charge débute par une anamnèse complète explorant les symptômes, la prise de médications, une enquête alimentaire, les voyages effectués, les expositions professionnelles et les hobbies, les antécédents personnels et familiaux.
L’examen clinique est soigneux et recherche toute atteinte d’organe cible.
Les examens complémentaires comprennent une biologie sanguine extensive (fonctions rénale et hépatique, ionogramme, troponine, vitamine B12 et tryptase (prédictifs d’une néoplasie myéloproliférative) (4), électrophorèse des protéines, bilan auto-immun (anticorps antinucléaires, ANCA), formule hémo-leucocytaire, frottis sanguin, sérologies parasitaires et virales, immunophénotypage lymphocytaire), une coproculture, un bilan cardiaque (ECG et surtout échographie transthoracique) (7), des épreuves fonctionnelles respiratoires et un scanner thoraco-abdominal.
En cas d’absence de cause secondaire au terme de ces explorationsune ponction-biopsie de moelle doit être réalisée avec immuno-histochimie, immuno-phénotypage, biologie moléculaire et cytogénétique. Ceci permet de différencier la leucémie chronique à éosinophiles, les néoplasies myéloprolifératives associées à une éosinophilie, le L-HES et enfin en cas d’absence de toute anomalie le I-HES en cas de lésion d’organe (1,2).
TRAITEMENT
En cas de syndrome hyperéosinophilique secondaire il s’agit bien évidemment d’en traiter la cause. Nous décrirons ici plus en détails la prise en charge des néoplasies myéloprolifératives associées à une éosinophilie, des L-HES et des I-HES.
Les patients atteints du variant myéloprolifératif, avec FIP1L1-PDGFR positif ou une mutation dans un des gènes PDGFRA ou PDGFRB, sont particulièrement sensibles à l’imatinib (un inhibiteur de tyrosine kinase à administration orale) et il s’agit du traitement de première ligne (6). L’évolution naturelle sans traitement est rapidement défavorable et mêmes les patients asymptomatiques au moment du diagnostic seront traités. En cas d’atteinte cardiaque une corticothérapie est associée pour une durée de 1-2 semaines (8).L’imatinib est poursuivi à vie au dosage minimal efficace. Les effets secondaires potentiels sont une neutropénie et des myalgies. En cas de résistance à l’imatinib une allogreffe peut être proposée.
Les patients atteints de mutations de FGFR1 font l’objet d’une chimiothérapie intensive puis d’une allogreffe en raison d’un très mauvais pronostic et d’une résistance à l’imatinib (1,2).
Les mutations de JAK2 (PCM1-JAK2) sont également de mauvais pronostic avec une évolution rapide vers une leucémie myéloblastique aigue et l’objectif est de proposer une allogreffe dans les meilleurs délais (2).
Pour les autres variants, L-HES et I-HES, le traitement initial consistera en l’administration de corticoïdes. La présence d’une strongyloïdose doit toujours être exclue au préalable en raison d’une possible évolution vers la dissémination et le décès. A noter que contrairement aux patients avec mutation FIP1L1-PDGFR, seuls les patients symptomatiques ou avec lésion d’organe sont traités, les autres bénéficient d’un suivi régulier (tous les 3-6 mois). La dose est adaptée à chaque patient et sera lentement dégressive.
Une fois la maladie stabilisée, une épargne des corticoïdes est préférable afin d’éviter leurs effets secondaires. L’hydroxyurée et l’interféron alpha sont fréquemment employés en seconde ligne de traitement. L’hydroxyurée est préférée pour la plupart des HES et présente peu d’effets secondaires (cytopénies ou intolérance digestive). L’interféron alpha peut être utilisé en bithérapie avec les corticoïdes pour les L-HES. Ses effets secondaires principaux sont un syndrome grippal, un état dépressif ou une asthénie.
Le mepolizumab (un anticorps humanisé IgG anti-IL-5) est actuellement proposé dans les études cliniques en 3ème ligne et donne des résultats prometteurs. Une étude multicentrique en double aveugle versus placebo ena analysé la tolérance et l’efficacité à long terme (9). Les effets secondaires sont mineurs (toux, asthénie, sinusite), il est donc généralement très bien toléré. Le mepolizumabpermet une épargne de corticoïdessans perte d’efficacité à long terme (63% des patients sont sous mepolizumab seul en fin d’étude) (9). Le taux d’éosinophiles est conservé 12 semaines après la dernière dose (10).
Enfin, en cas de résistance aux différents agents, une allogreffe est également possible.
CONCLUSION
Le syndrome hyperéosinophilique est une pathologie rare dont la définition et la physiopathologie ont évolué au fil des progrès techniques. Des sous-groupes appelés variants ont été identifiés et permettent de proposer des thérapies ciblées, en particulier l’utilisation de l’imatinib pour les variants myéloprolifératifs (avec une grande sensibilité chez les FIP1L1-PDGFR positifs). Le développement de nouveaux traitements comme les anticorps anti-IL-5 a permis de réduire les effets secondaires des corticoïdes et d’améliorer la qualité de vie de ces patients.
AFFILIATIONS
1 Assistante en Médecine Interne
2 Service de Maladies Infectieuses, CHL, Luxembourg
3 Service d’Hématologie-Oncologie, CHL, Luxembourg
CORRESPONDANCE
Dr. LÉA SCHMITZ
Médecine interne
lea.schmitz@student.uliege.be
Dr. LAURENT PLAWNY
Hématologie-Oncologie
plawny.laurent@chl.lu
Centre Hospitalier de Luxembourg
Rue Ernest Barblé 4
L-1210 Luxembourg
RÉFÉRENCES
- Gotlib J. - CME Information: World Health Organization-defined eosinophilic disorders: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015 Nov; 90(11):1077-89. doi: 10.1002/ajh.24196. Ouvrir dans PubMed
- Reiter A, Gotlib J. Myeloid neoplasms with eosinophilia. Blood. 2017; 129(6) : 704-714. Ouvrir dans PubMed
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012; 130 : 607. Ouvrir dans PubMed
- Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015; 126(9):1069–1077. Ouvrir dans PubMed
- Curtis C, Ogbogu P. Hypereosinophilic Syndrome.Clinic Rev Allerg Immunol. 2016; 50:240–251. Ouvrir dans PubMed
- Savage N, George TI, Gotlib J. Myeloid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, and FGFR1: a review. Int. Jnl Lab Hem. 2013; 35: 491–500. Ouvrir dans PubMed
- Jin X, Ma C, Liu S, et al. Cardiac involvements in hypereosinophilia-associated syndrome: Case reports and a little review of the literature. Echocardiography. 2017; 00: 1–5.
- Pitini V, Arrigo C, Azzarello D, et al. Serum concentration of cardiac Troponin T in patients with hypereosinophilic syndrome treated with imatinib is predictive of adverse outcomes. Blood. 2003, 102 : 3456. Ouvrir dans PubMed
- Roufosse FE, Kahn JE, Gleich GJ, et al. Long-term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J Allergy Clin Immunol. 2013, 131(2): 461–467. Ouvrir dans PubMed
- Menzella F, Lusuardi M, Galeone C, et al. - Profile of anti-IL-5 mAb mepolizumab in the treatment of severe refractory asthma and hypereosinophilic diseases. J Asthma Allergy. 2015 Oct 8;8:105-14. doi: 10.2147/JAA.S40244. Ouvrir dans PubMed