Introduction
Il s’agit d’une pathologie auto-immune dans laquelle des auto-anticorps sont dirigés contre les phospholipides membranaires. Les principales manifestations sont des évènements thrombotiques ou thrombo-emboliques divers exposant les patients à une morbidité importante par dysfonction organique. Des progrès thérapeutiques sont en cours et de nouvelles cibles sont à l’étude mais actuellement l’anticoagulation par anti-vitaminique K (AVK) reste le traitement de référence en dehors des grossesses.
Nous relatons le cas d’une jeune patiente présentant une atteinte multi-organique étalée sur une période de plusieurs semaines avec un tableau clinique complexe et chez qui l’initiation d’un traitement anticoagulant dès l’apparition de la symptomatologie est primordial.
Cas clinique
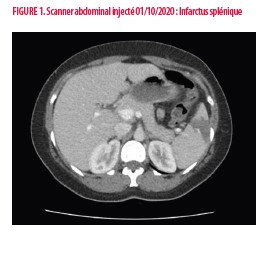
Il s’agit d’une patiente de sexe féminin âgée de 43 ans, non tabagique, sans co-morbidités notables et sans traitement chronique. Trois grossesses ont été menées à terme sans complication. Dans le mois précédent son admission, elle a bénéficié d’une myomectomie hystéroscopique, interrompue en raison de complications hémorragiques. Elle se présente aux urgences pour vomissements et douleur abdominale. Cliniquement, présence d'une défense en région épigastrique et hypochondre gauche. Les paramètres hémodynamiques sont stables et la biologie de base rassurante. Le Scanner abdominal objective un infarcissement splénique (Figure 1). Un complément biologique est demandé en salle d’urgences, avec notamment la recherche d’anticorps antiphospholipides.
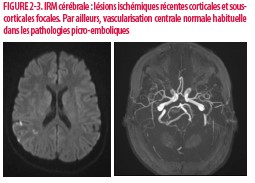
Cinq jours plus tard, le diagnostic échographique de thrombophlébite superficielle d’un membre supérieur à l’emplacement d’un cathéter périphérique est posé. Cet évènement est rapidement suivi d’une symptomatologie d’AIT sous forme de troubles visuels transitoires menant à la réalisation d’une IRM cérébrale objectivant des lésions vasculaires ischémiques pariétales droites d’origine emboligène (Figures 2-3). L’Échographie doppler des vaisseaux du cou et l’échocardiographie sont normales. Devant ce tableau clinique, un bilan vasculaire étiologique classique négatif et à la lumière des résultats biologiques effectués en salles d’urgence (anticoagulants lupiques et anticorps anti bêta 2 glycoproteines positifs), le diagnostic de SAPL est posé et le traitement anticoagulant est initié (héparine de bas poids moléculaire (HBPM) puis AVK).
Discussion
Le syndrome des anticorps antiphospholipides (SAPL) est une maladie systémique auto-immune dans laquelle des auto-anticorps sont dirigés contre les phospholipides membranaires (1,2). Il est décrit comme primaire, si présent de façon isolée, ou secondaire s’il est retrouvé en association à d’autres maladies auto-immunes (comme le lupus érythémateux systémique ou la polyarthrite rhumatoïde) (2-5).
On considère qu’environ 5% à 10% de la population jeune présente des anticorps antiphospholipides, mais une minorité développera un syndrome des anticorps antiphospholipides. Certains facteurs intrinsèques ou extrinsèques semblent accentuer le risque de développer un SAPL tels que des facteurs génétiques (thrombophilie constitutionnelle), des infections (borrélia burgdorferi, tréponème, VIH, leptospirose), ou la prise de certains médicaments (6).
Ce syndrome associe nécessairement la présence de marqueurs biologiques et la présence de manifestations cliniques. Les critères diagnostiques de Sapporo élaborés au Japon en 1999 ont été mis à jour en 2006 à Sydney (7,8).
1. À la biologie, trois types d’anticorps antiphospholipides peuvent être retrouvés : l’anticoagulant lupique, l’anticorps anti-β2-glycoprotéine-1 et les anticorps anticardiolipines. L’anticoagulant du lupus a été découvert chez les patients atteint d’un lupus érythémateux systémique. Il a été appelé « anticoagulant » à la suite de son action in vitro mais il est bien pro-coagulant in vivo (2). Les recommandations précisent que les anticorps doivent être présents à deux reprises lors de tests de laboratoire réalisés à minimum 12 semaines d’intervalle (1-3,7).
2. Les manifestations cliniques associant des phénomènes thrombotiques récurrents tant au niveau veineux qu’artériels et/ou une complication obstétricale (7).
Au niveau thrombotique : tout le territoire vasculaire est à risque d’être entrepris (8).
a) Veineux : TVP et TVS principalement localisées au niveau des membres inférieurs.
b) Artériel : L’accident vasculaire cérébral est le plus fréquent (1,3,6). D’autres localisations sont décrites comme chez notre patiente où un infarcissement splénique a été la première manifestation clinique de la maladie. L’occlusion de l’artère centrale de la rétine, des microangiopathies répétées au niveau cérébral ou rénal menant à des dysfonctions chroniques sont décrites (6). Des thromboses plus proximales, notamment au niveau de l’artère rénale donnant lieu à une hypertension artérielle réfractaire sont également retrouvées (6).
Au niveau obstétrical : Mort in utéro (à plus de 10 semaines de gestation), grande prématurité, présence de 3 fausses-couches spontanées précoces inexpliquées (1-6), autres (pré-éclampsie, détresse fœtale aigue, retard de développement in-utérin, insuffisance placentaire, et HELLP syndrome (1)).
3. Certains patients (moins de 1%) peuvent développer une forme de SAPL dite « catastrophique » dans laquelle de nombreux phénomènes thrombotiques se développent simultanément dans différents organes ou tissus (1,6). Une confirmation histopathologique de phénomènes thrombotiques et la présence d’anticorps antiphospholipides est également nécessaire au diagnostic (1,6). Ces phénomènes de thromboses généralisées mènent rapidement à une défaillance multi-organique et sont associés à une mortalité élevée. Différents facteurs précipitants sont décrits (infection, chirurgie, contraceptif oral, complication obstétricale, arrêt d’un traitement anticoagulant, cancer) (1,3,4).
4. D’autres manifestations plus rares existent : thrombocytopénie (15% des patients), livedo reticularis, ulcère de peau, valvulopathie cardiaque (Libman-Sacks), chorée (1,4). Ce sont le plus souvent les valves aortiques ou mitrales qui sont touchées, pouvant mener à une insuffisance ou à une sténose valvulaire (6).
La physiopathologie du SAPL n’est pas totalement élucidée. La présence d’anticorps antiphospholipides est décrite comme nécessaire mais non suffisante au développement de thrombus. Un élément déclencheur pro-thrombotique (trigger) est nécessaire (infection, lésion endothéliale, inflammation, facteurs immunologiques, pise d’oestro-progestatif, chirurgie ou immobilisation) (2,6). La pathogénie des anticorps antiphospholipides a été décrite selon plusieurs mécanismes avec notamment l’activation de récepteurs à la surface des cellules endothéliales, des monocytes et des plaquettes (1,2). L’activation de différentes voies de signalisation via ces anticorps mène à l’expression de molécules d’adhésion et de facteurs tissulaires, entrainant un état inflammatoire et pro-coagulant. De plus, il semblerait que l’activation de la voie du complément jouerait un rôle important dans la physiopathologie de cette maladie, surtout dans la survenue de complications obstétricales. (1,2).
Une prophylaxie primaire, par aspirine à dose de 75 à 100mg, est discutable chez les patients présentant des anticorps antiphospholipides (8,9). Celle-ci est d’autant plus indiquée s’ils cumulent d’autres facteurs de risque thrombo-emboliques (3-5).
Le traitement recommandé reste actuellement l’anticoagulation des patients par AVK avec un INR visé habituellement entre 2 et 3 (1-3,5,9). Une surveillance étroite des patients est nécessaire suite notamment aux interactions médicamenteuses, diététiques, liées aux maladies intercurrentes, au tabagisme, à la prise d’alcool ou à l’exercice sportif (3). Il est recommandé que le traitement soit maintenu à vie (2,3,5,8). Le traitement anticoagulant peut être exceptionnellement interrompu, sur avis d’expert, lors d’un épisode unique de thrombose veineuse favorisée par un facteur extrinsèque contrôlé (4,9).
Chez les patients ayant présenté une thrombose artérielle, et notamment une thrombose cérébrale, les recommandations restent incertaines (AVK versus AVK+aspirine à faible dose) (3,5,9). Certaines études soulignent le risque majoré de saignement lors de l’association d’antiagrégant et d’anticoagulant et préconisent un traitement par AVK ou par clopidogrel (4).
D’autres études suggèrent l’utilisation d’AVK seul avec un INR cible entre 3 et 4 (9).
Les anticoagulants oraux directs (AOD) dans le SAPL sont discutés mais, à ce jour, le manque d’études cliniques significatives et de résultats positifs ne permet pas de les utiliser de façon systématique en pratique (2). Certaines études ont montré une majoration des évènements thrombotiques récurrents sous AOD (8-9). Dans les cas des SAPL avec thromboses veineuses, les anticoagulants oraux directs pourraient, actuellement, être prescrits en cas d’allergie, d'intolérance aux AVK ou chez les patients avec un mauvais contrôle de l’anticoagulation (2-9).
Lors des grossesses, les patientes ayant des antécédents thrombotiques sont traitées par une association d’aspirine à faible dose et d’HBPM à dose thérapeutique (1-4-5-9). Chez les patientes présentant uniquement les complications obstétricales du SAPL, il est recommandé que le traitement par aspirine et HBPM à dose prophylactique soit maintenu jusqu’à 6 semaines après l’accouchement (2,4,5,8,9).
De plus, il faudra toujours identifier et traiter les autres facteurs de risque cardio-vasculaires/thrombogènes tels que l’hyperlipidémie, le tabagisme, l’hypertension artérielle, la prise d’œstrogènes, l’immobilisation, etc. (1,3,9).
Malgré les traitements anticoagulants bien menés, il persiste un risque de récidive thromboembolique, entrainant une morbi-mortalité non négligeable. La compréhension progressive de la physiopathologie, son caractère pro-inflammatoire et notamment l’implication de l’activation de la voie du complément permet d’envisager de nouvelles pistes thérapeutiques (1,8). Des traitements par hydroxychloroquine, rituximab, statine, sirolimus ou plus spécifiquement éculizumab (anticorps monoclonal dirigé contre la fraction C5 du complément) sont en cours d’évaluation. Ils représentent une cible de traitement attractive (1,2,3,8).
Conclusion
Notre patiente a présenté une forme de SAPL rapidement évolutive avec plusieurs évènements thrombo-emboliques rapprochés. Il est essentiel d’évoquer un diagnostic de SAPL devant ce tableau clinique et d’initier rapidement le traitement anticoagulant par AVK.
Le syndrome des anticorps antiphospholipides est une pathologie rare qu’il est important de connaitre et de pouvoir évoquer notamment dès la prise en charge en salle d’urgences. Si la survie à 10 ans est très bonne dans les études européennes, la morbidité de la maladie reste élevée avec notamment le développement d’insuffisance chronique multi-organique, d’hypertension artérielle pulmonaire, de gangrène des extrémités… Il est dès lors crucial de veiller à prendre en charge ces patients dans leur globalité et avec une participation interdisciplinaire (6).
Affiliations
1. Cliniques universitaires Saint-Luc, Urgences, B-1200 Bruxelles
2. Cardiologue Urgentiste (MD, FESC) : Département des Urgences, Clinique Notre-Dame de Grâce, Gosselies, Belgique
3. Chef de service adjoint des Urgences, Département des Urgences, Clinique Notre-Dame de Grâce, Gosselies, Belgique
4. Département des Urgences et des soins intensifs, Clinique Notre-Dame de Grâce, Gosselies, Belgique
5. Cheffe de service, Département des Urgences, Clinique Notre-Dame de Grâce, Gosselies, Belgique
Correspondance
Dr. Camille Desender
Université catholique de Louvain
Cliniques universitaires Saint-Luc
Urgences
Avenue Hippocrate 10
B-1200 Bruxelles
camille.desender@student.uclouvain.be
Références
- Shruti Chaturvedi and Keith R McCrae. Diagnosis and management of the antiphospholipid syndrome. Blood Rev. 2017 Nov; 31(6): 406–417.
- Deepa R.J., Arachchillage and Mike Laffan. Pathogenesis and management of antiphospholipid syndrome. Br J Haematol. 2017; 178: 181–195.
- Amine Ghembaza and David Saadoun. Management of Antiphospholipid Syndrome. Biomedicines. 2020; 8, 508.
- M. Limper, K. de Leeuw, A.T. Lely, et al. Diagnosing and treating antiphospholipid syndrome: a consensus paper. Nether J Med. 2019 Apr;77(3):98-108.
- Wendy Lim, MD; Mark A. Crowther, MD; John W. Eikelboom, MBBS. Management of Antiphospholipid Antibody Syndrome A Systematic Review. JAMA. 2006; 295(9): 1050-1057.
- Jean G. Bustamante, Amandeep Goyal, Pankaj Bansal, et al. Antiphospholipid Syndrome. StatPearls. Last Update: July 4, 2022.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006; 4: 295–306.
- Mia Rodziewicz and David P. D’Cruz. An update on the management of antiphospholipid syndrome. Ther Adv Musculoskelet Dis. 2020; 12: 1759720X20910855.
- Tektonidou MG, Andreoli L, Limper M, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. 2019; 78: 1296–1304.
- Tektonidou MG, Andreoli L, Limper M, et al. Management of thrombotic and obstetric antiphospholipid syndrome: a systematic literature review informing the EULAR recommendations for the management of antiphospholipid syndrome in adults. RMD Open. 2019; 5: e000924.
- David A. Garcia, Munther A. Khamashta, Mark A. Crowther. How we diagnose and treat thrombotic manifestations of the antiphospholipid syndrome: a case-based review. Blood. 2007; 110 (9): 3122–3127.
- R A Asherson 1, R Cervera, P G de Groot, et al. Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12(7):530-4.