MANIFESTATIONS DERMATOLOGIQUES DU LUPUS ÉRYTHÉMATEUX ET DE LA DERMATOMYOSITE
C. Francès
LE LUPUS ÉRYTHÉMATEUX
Le Professeur C. Francès rappelle que les manifestations dermatologiques du lupus érythémateux (LE) sont classées en trois catégories : les lésions lupiques ou spécifiques, les lésions vasculaires et les lésions ni lupiques ni vasculaires.
1. Les lésions lupiques ou spécifiques du LE
Clinique
Les lésions lupiques peuvent être aiguës, subaiguës ou chroniques.
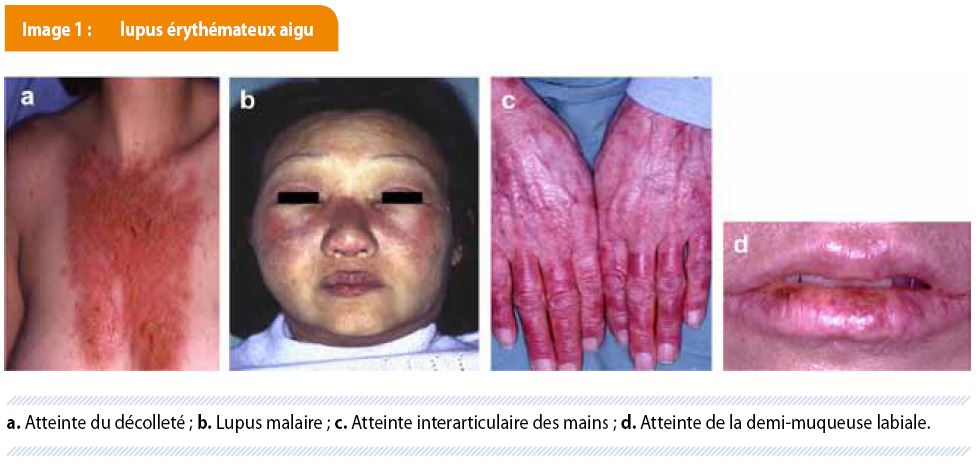
Le lupus érythémateux aigu (LEA) touche surtout les femmes en âge de procréer (85 % des cas). L’aspect clinique associe des lésions érythémateuses mal limitées, de l’œdème, des squames, et parfois, des bulles et des érosions. Les lésions sont classiquement situées au niveau des zones exposées, réalisant notamment la topographie dite « en masque de loup » au niveau du visage (lupus malaire) (Image 1 a et b). On peut également retrouver une atteinte des zones interarticulaires des mains (Image 1c) et de la muqueuse buccale. En effet, l’atteinte érosive de la demi-muqueuse labiale est très fréquente dans le LEA (Image 1d). Ces lésions buccales érosives sont statistiquement très souvent associées à une atteinte rénale. Il existe aussi des formes diffuses de LEA, de type morbilliforme. Dans tous les cas, le traitement du LEA entraine une disparition des lésions sans cicatrices.
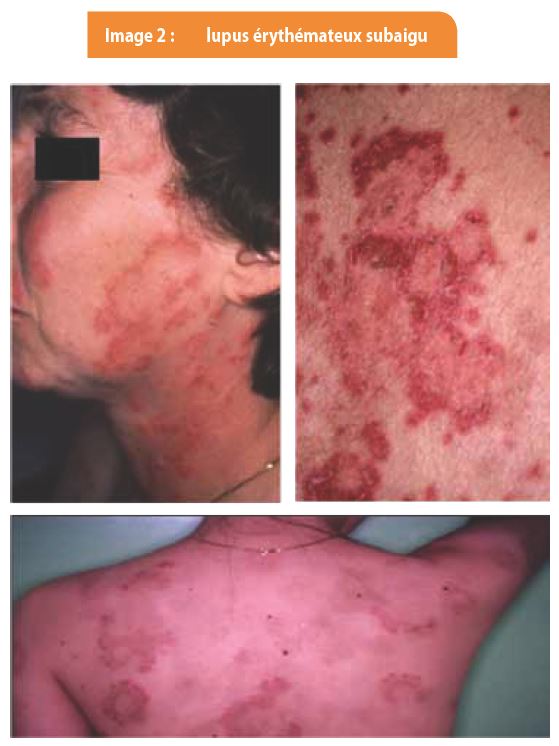
Le lupus érythémateux subaigu (LESA) touche essentiellement les femmes caucasiennes. C’est cette forme de lupus que l’on retrouve dans le lupus néonatal, dans le lupus associé à un déficit en complément et dans les lupus induits par des médicaments. Il se présente sous forme de plaques annulaires, de lésions psoriasiformes et/ou de lésions érosives (Image 2). Il touche habituellement la moitié supérieure du corps, surtout les zones exposées et la cavité buccale. Les lésions érosives buccales ne sont par ailleurs pas associées à une atteinte rénale, contrairement au LEA. Les lésions de LESA peuvent laisser des séquelles pigmentaires.
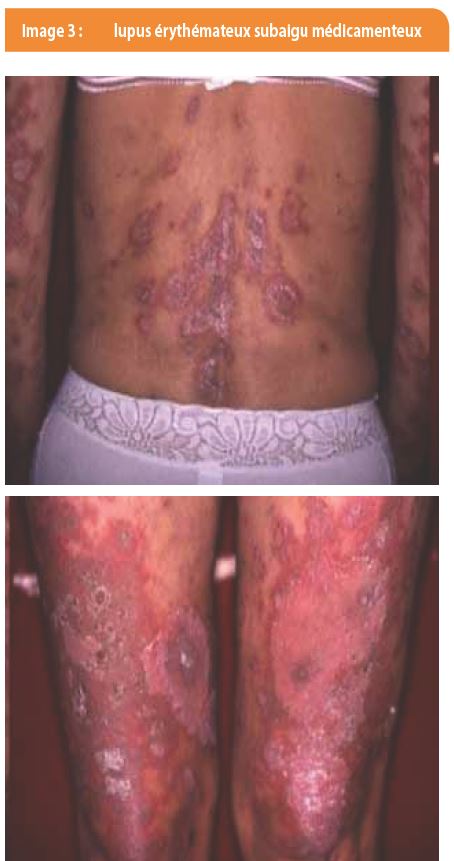
Le LESA peut être d’origine médicamenteuse. Il est alors induit par des médicaments pris depuis plusieurs semaines voire des années. Il se résout en plusieurs semaines ou mois après arrêt du traitement inducteur mais les anticorps anti-SSA/Ro persistent après résolution de l’éruption. En effet, le médicament ne joue qu’un rôle déclencheur chez des sujets possédant des caractéristiques génétiques et sérologiques (anticorps anti-SSA/Ro) particulières. Les principaux médicaments inducteurs sont par ordre de fréquence décroissante, la terbinafine, les anti-TNF, les anti-épileptiques et les inhibiteurs de la pompe à protons. Dans les formes induites, les lésions sont souvent plus étendues (Image 3). La présence de lésions dans des localisations inhabituelles, notamment au niveau de la moitié inférieure du corps, doit faire rechercher une cause médicamenteuse.
Les différentes formes de lupus érythémateux chronique (LEC) sont le lupus discoïde, le lupus-engelure, la panniculite lupique et le lupus tumidus ou intermédiaire.
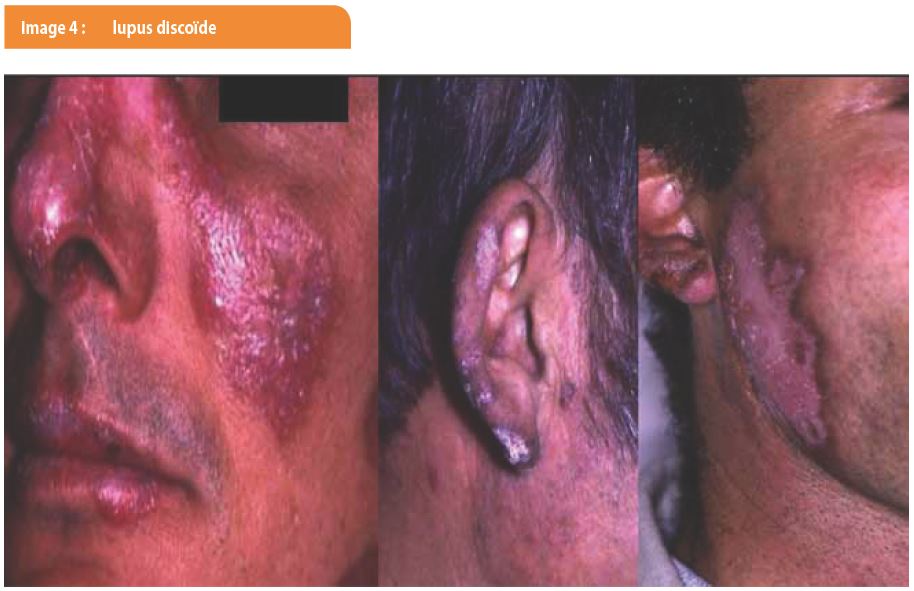
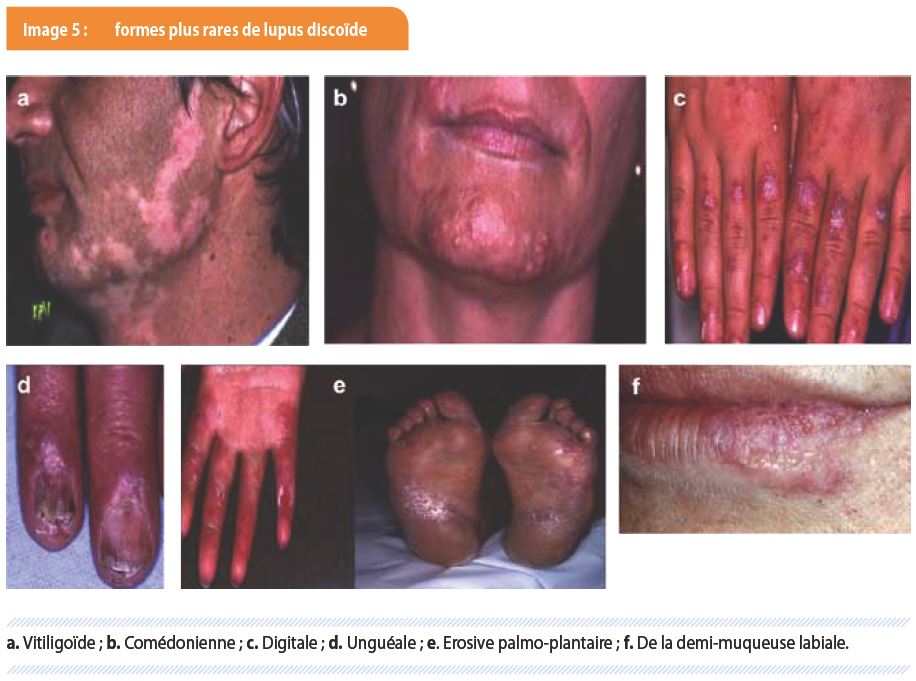
Le lupus discoïde (LD) touche surtout les adultes entre 20 et 40 ans, majoritairement des fumeurs et, dans 60 % des cas, des femmes. Il peut être localisé à la région céphalique, disséminé, ou encore buccal. Les lésions cliniques associent érythème, hyperkératose folliculaire et atrophie cicatricielle définitive (Image 4). De la sorte, il constitue une urgence esthétique en raison de l’atrophie permanente qu’il provoque. Dans sa forme céphalique, les lésions prédominent au niveau des joues, des oreilles et du cuir chevelu. Dans sa forme buccale, les lésions sont proches de celles du lichen plan. Des formes plus rares existent également, notamment, vitiligoïde, comédonienne, digitale, unguéale, érosive palmo-plantaire et celle de la demi-muqueuse labiale (Image 5).
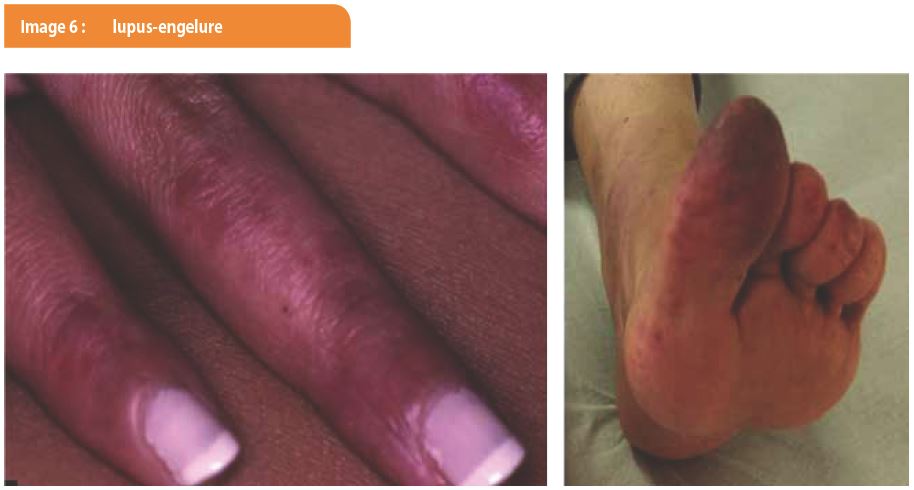
Le lupus-engelure se caractérise par des engelures persistantes au-delà de la saison froide (Image 6). Il touche classiquement les doigts et les orteils. Le diagnostic est guidé par l’histologie lupique et l’évolution clinique des lésions.
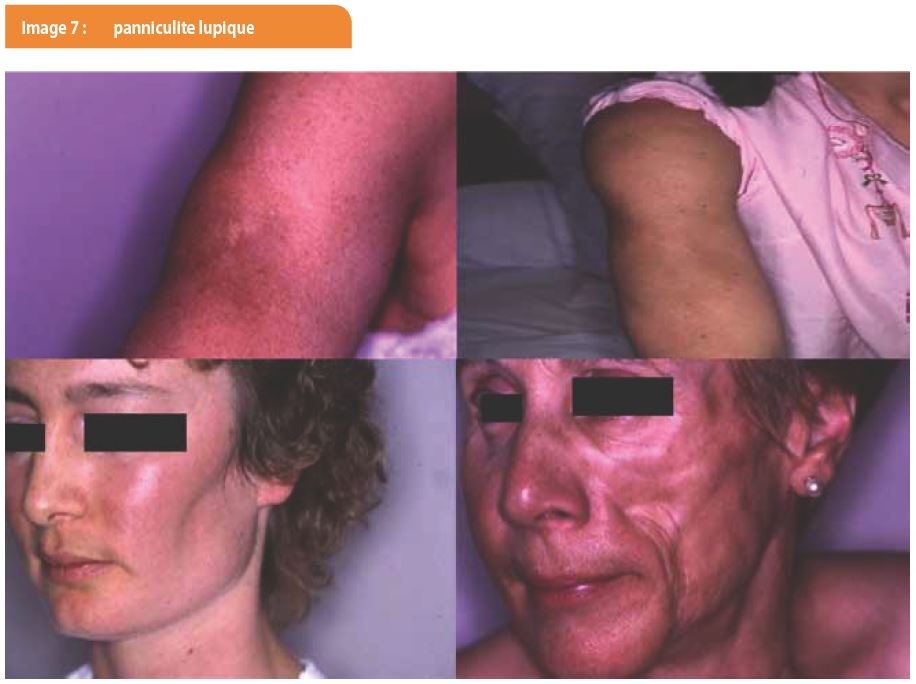
La panniculite lupique se manifeste par des placards indolores infiltrés, pouvant s’ulcérer et se nécroser. Après plusieurs années, ils évoluent vers des cicatrices atrophiques inesthétiques et définitives, parfois calcifiées (Image 7). Les zones atteintes sont les régions deltoïdiennes, les joues, les cuisses et les seins. A l’histologie, on ne retrouve pas de dermite d’interface. Le principal diagnostic différentiel est la panniculite histiocytaire cytophagique.
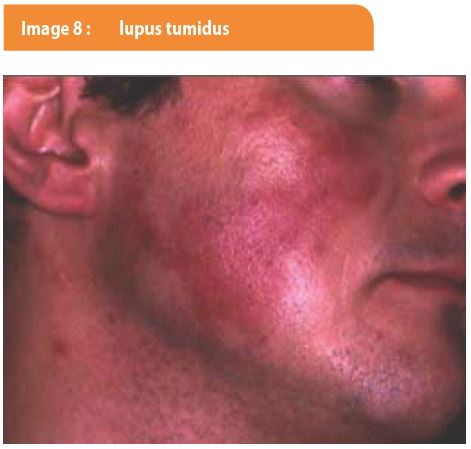
Le lupus tumidus (LT) ou intermédiaire se manifeste par des placards saillants, de teinte rouge, à limites nettes (Image 8). Il atteint essentiellement le visage et le tronc. Il n’y a pas d’hyperkératose folliculaire, ni d’atrophie résiduelle et est ainsi considéré comme la forme la moins sévère de LE. Il présente un spectre continu avec la maladie de Jessner-Kanoff (infiltrat lymphocytaire cutané bénin dont l’individualité est toujours controversée mais qui se rapprocherait d’une forme cutanée pure de LE).
Diagnostic
Le diagnostic de lupus cutané est essentiellement clinique et histologique. Il se fait sur base de l’aspect clinique évocateur, de l’histologie et de l’évolution. Les caractéristiques anatomopathologiques communes aux trois formes de lupus érythémateux sont une dermite d’interface non spécifique et non constante avec une atrophie épidermique, une atteinte des kératinocytes basaux, un épaississement de la membrane basale et un infiltrat lymphocytaire du derme. Les trois formes se différencient les unes des autres par le degré d’hyperkératose, l’importance de l’œdème dermique et la topographie et la densité de l’infiltrat. L’immunofluorescence cutanée en peau lupique a quant-à-elle un intérêt diagnostic mineur. En effet, on retrouve des dépôts d’immunoglobulines et/ou de complément à la jonction dermo-épidermique dans 90 % des LEA, 60 % des LESA et 90 % des LD, mais en aucun cas la négativité de l’immunofluorescence ne permet d’éliminer le diagnostic de maladie lupique.
Parmi les diagnostics différentiels du lupus érythémateux cutané, on retrouve :
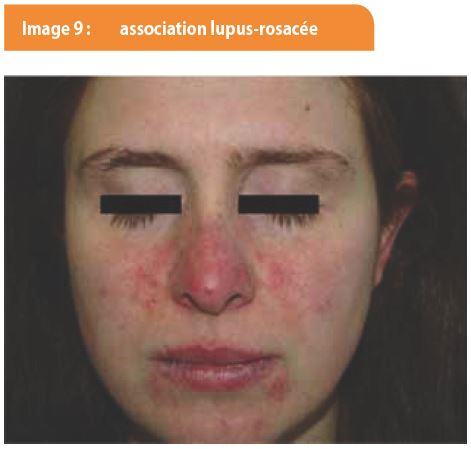
1. La rosacée. Les deux principaux éléments la différenciant du LE sont la présence de pustules et la topographie des lésions. En effet, des pustules sont classiquement retrouvées dans les lésions de rosacée mais sont absentes des lésions lupiques. De plus, on peut retrouver une topographie « en masque de loup » dans la rosacée mais d’autres zones sont habituellement également atteintes, à savoir le front, les yeux et le menton. Cependant, le diagnostic est parfois plus difficile. Il existe des formes d’association lupus-rosacée, avec une clinique de rosacée au niveau des joues associée à une atteinte de la demi-muqueuse des lèvres de type lupique (Image 9). Ces formes sont assimilées au lupus tumidus car d’évolution favorable.
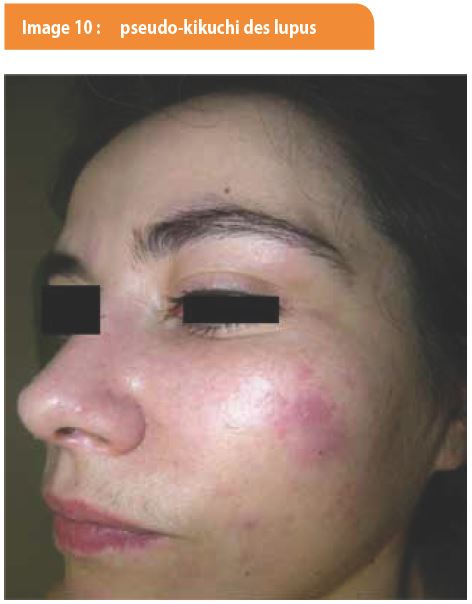
2. Le Pseudo-kikuchi des lupus. La maladie de Kikuchi est une affection bénigne rare qui se caractérise par l’apparition d’adénopathies douloureuses, généralement accompagnée de fièvre modérée et de sueurs nocturnes, et qui se définit par une histologie ganglionnaire typique. On y retrouve une atteinte cutanée dans 30 à 60 % des cas. Dans le pseudo-Kikuchi, l’atteinte est exclusivement cutanée sans implication ganglionnaire, mais l’histologie cutanée a les mêmes caractéristiques que celles de l’histologie ganglionnaire de Kikuchi. On a donc surnommé cette pathologie le pseudo-Kikuchi des lupus car elle est responsable d’une atteinte cutanée pure de type lupique sous forme de plaques papuleuses ou annulaires, sans atteinte ganglionnaire, mais avec une histologie dite de Kikuchi (Image 10). En effet, contrairement au lupus cutané, on a certes une dermite de jonction dans 50 % des cas mais on retrouve un infiltrat dermique essentiellement interstitiel et de composition tout à fait différente (infiltrat mononucléé histiocytaire avec noyau en croissant ne contenant pas de polynucléaires neutrophiles).
Pour ce qui est de la place des manifestations cutanées lupiques dans le spectre de la maladie lupique, le Professeur Francès précise que :
- pour le LEA, plus de 90 % ont ou auront un lupus systémique (LS).
- pour le LESA, 50 % ont un LS et 7 à 21 % des LS ont des lésions de LESA.
- pour le LD, 10 à 20 % ont ou auront un LS et 15 % des LS ont des lésions de LD.
Il est intéressant de relever que dans les critères diagnostiques du lupus érythémateux systémique de l’Association de Rhumatologie Américaine, quatre des onze critères sont dermatologiques, à savoir l’érythème malaire, le lupus discoïde, la photosensibilité et les ulcérations orales. Par ailleurs, il est important de noter que la présence d’anticorps anti-noyaux ou une atteinte hématologique suffisent à définir la pathologie comme systémique alors qu’il n’y a pas d’atteinte viscérale.
Traitement
Le traitement des lésions lupiques cutanées comprend avant tout une protection solaire, associée à une supplémentation en vitamine D (400 UI de vitamine D3/jour).
Parmi les traitements locaux, les dermocorticoïdes sont très efficaces mais du fait de leurs effets secondaires potentiels (atrophie cutanée et dermite des corticoïdes), ils sont à éviter sur une longue période (> 6 semaines) au niveau du visage. Les immunomodulateurs topiques (tacrolimus à 0,1 % (Protopic®) et pimécrolimus à 1 % (Elidel®)) sont surtout efficaces sur les lésions de LT, de LESA et de LD, et d’autant plus que les lésions sont récentes. Les rétinoïdes locaux (tazarotène et trétinoïne) donnent des résultats uniquement sur les LD très hyperkératosiques.
En ce qui concerne les traitements systémiques, le traitement de première intention reste les antipaludéens de synthèse (APS), avec une préférence pour l’hydroxychloroquine par rapport à la chloroquine car responsable de moins d’effets secondaires oculaires. La toxicité oculaire est directement liée à la dose (en mg/kg). Les doses recommandées sont respectivement de 6,5 mg/kg/jour et de 4 mg/kg/jour pour l’hydroxychloroquine et la chloroquine. Leur efficacité est jugée à trois mois de traitement et une amélioration est notée dans 50 à 70 % des cas. La quinacrine, APS anciennement utilisé, sans toxicité oculaire, n’est plus disponible ni en France ni en Belgique.
En cas de suspicion de lupus cutané résistant aux APS, il faut tout d’abord s’assurer du diagnostic et d’une prise médicamenteuse correcte par dosage sanguin de l’hydroxychloroquine. En effet, cette dernière peut causer des nausées et un goût amer, incitant ainsi le patient à négliger le traitement. Si le diagnostic est confirmé et que le taux sanguin est adéquat, on doit alors changer d’APS (« shift » pour la chloroquine, plus efficace mais plus toxique) ou les associer. Si malgré ce « shift » ou cette association, le patient reste symptomatique, on parle de lupus cutané réfractaire aux APS. Parmi les alternatives thérapeutiques, on retrouve les médicaments suivant : le thalidomide, le lénalidomide, le méthotrexate, la disulone, les rétinoïdes, la sulfasalazine (si acétyleurs rapides), l’azathioprine, la ciclosporine ou le mycophénolate mofétil. Il faut impérativement éviter de recourir aux corticoïdes systémiques comme traitement des lésions cutanées du lupus car ils ne sont efficaces qu’à la dose d’1,5 mg/kg/jour, dose qui ne peut en aucun cas être maintenue au long court.
En cas de lupus cutané réfractaire aux APS, les tendances thérapeutiques divergent selon les pays, en particulier entre la France et les Etats-Unis. En France, on ajoute d’emblée le thalidomide à l’hydroxychloroquine. Si la résistance persiste, on remplace le thalidomide par le lénalidomide, toujours en maintenant l’association à l’hydroxychloroquine. En effet, le thalidomide comme le lénalidomide sont thrombogènes et, en plus d’une association à la prise d’aspirine, les propriétés antiagrégantes de l’hydroxychloroquine ont un effet favorable. Aux Etats-Unis, on utilise d’abord la quinacrine en association avec de l’hydroxychloroquine ou de la chloroquine et ce n’est qu’en cas d’inefficacité de ces associations que l’on recoure au méthotrexate, puis à la disulone et enfin au thalidomide.
2. Les lésions vasculaires
Les lésions vasculaires ne sont pas spécifiques du lupus érythémateux.
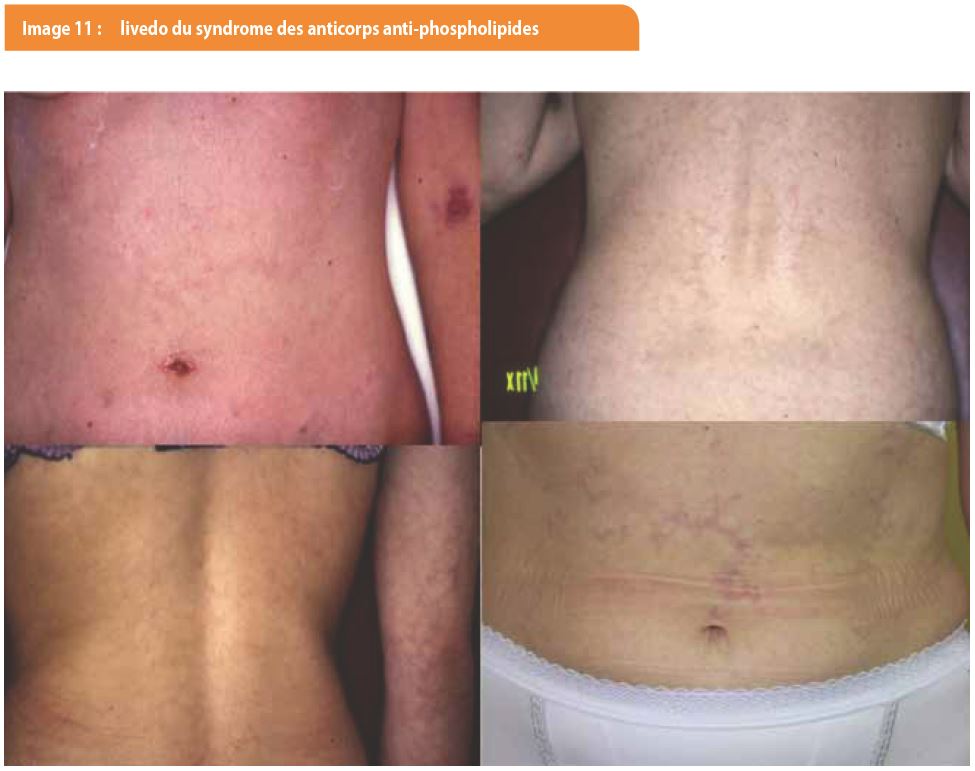
Le phénomène de Raynaud est retrouvé chez 10 à 45 % des patients atteints de lupus systémiques. L’urticaire est présente dans 4 à 13 % des lupus systémiques et peut être classique, neutrophilique ou liée à une vasculite leucocytoclasique des vaisseaux dermiques. Le livedo du syndrome des anticorps anti-phospholipides (SAPL) est un livedo ramifié, qu’il faut surtout rechercher au niveau du tronc, localisation où le livedo est pure et non falsifié par le livedo physiologique, contrairement à celui localisé aux cuisses et bras (Image 11). L’anatomopathologie d’un livedo n’a aucun intérêt en pratique sauf si on veut éliminer une vascularite. Parmi les autres lésions vasculaires, on retrouve parfois un purpura, des ulcères de jambes, des nécroses cutanées extensives, des hémorragies sous-unguéales multiples, un érythème palmaire et des télangiectasies péri-unguéales.
3. Les lésions ni lupiques ni vasculaires
Enfin, les lésions ni lupiques ni vasculaires parfois associées au LE sont l’effluvium télogène, les mucinoses en plaques et les anétodermies. A nouveau, ces lésions peuvent être associées au LE mais ne sont nullement spécifiques de celui-ci.
LA DERMATOMYOSITE
Les manifestations dermatologiques de la dermatomyosite (DM) sont également classées en trois catégories : les lésions spécifiques de DM, les lésions vasculaires et les lésions non spécifiques non vasculaires.
1. Les lésions spécifiques de dermatomyosite
Clinique
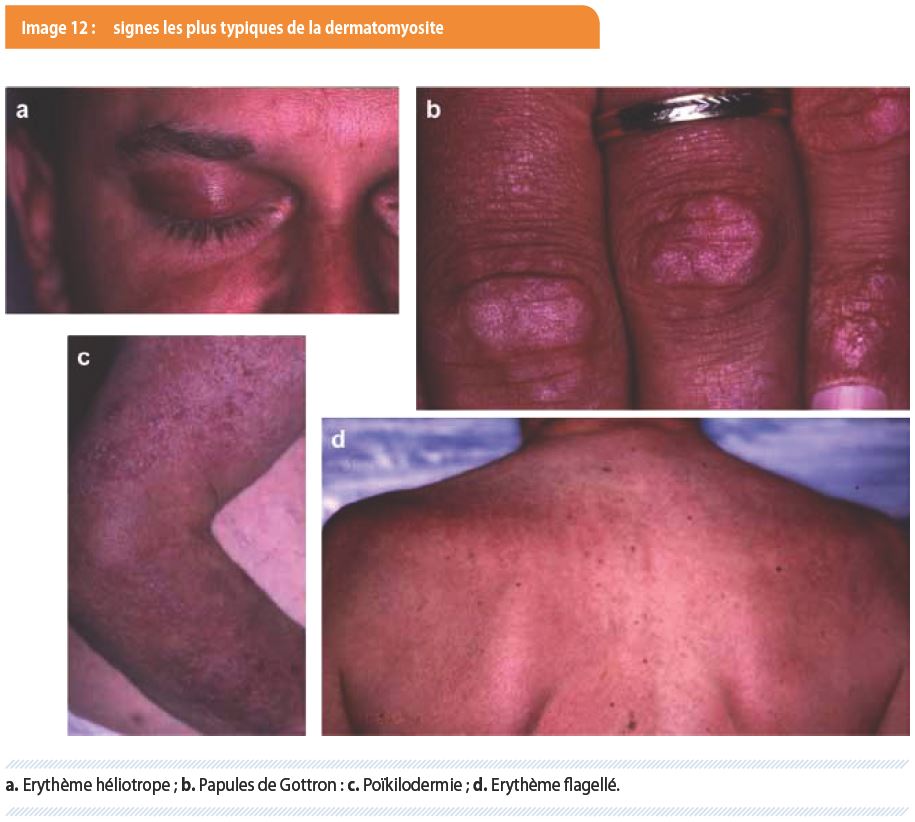
Les lésions spécifiques de DM prédominent sur les zones exposées (visage et mains). Le rôle de l’exposition solaire est reconnu dans 20 % des cas. Mais fréquemment, les lésions sont plus étendues. L’aspect élémentaire associe érythème, œdème et kératose. L’image 12 reprend les signes les plus typiques de la DM, à savoir :
- l’érythème héliotrope rose lilacé des paupières supérieures ;
- les papules de Gottron, lésions érythémato-papuleuses localisées au niveau des articulations métacarpo-phalangiennes et inter-phalangiennes des mains ;
- la poïkilodermie, plus diffuse, associant atrophie, télangiectasies et leucomélanodermie ;
- l’érythème flagellé du haut du dos.
Des formes atrophiques parfois ulcérées et des lésions vésiculo-bulleuses, ulcérées et nécrotiques sont également décrites. L’association nécrose cutanée et néoplasie est connue de longue date, la nécrose ayant une valeur prédictive positive pour un cancer de 85 % et une sensibilité de 58 %.
Histologie
L’anatomopathologie des lésions spécifiques montre une dermite d’interface avec vacuolisation et nécrose des kératinocytes basaux, un atrophie du corps muqueux, une hyperkératose orthokératosique, un œdème dermique superficiel et infiltrat lymphocytaire CD4 périvasculaire. La présence d’une vascularite histologique est statistiquement en faveur d’une DM associée à une néoplasie. A l’immunofluorescence directe, on peut retrouver une bande lupique, des dépôts de C3 ou d’IgG à la jonction dermo-épidermique dans 10 à 20 % des DM et la présence du complexe membranaire d’attaque C5b-9 le long de la jonction dermo-épidermique ou des vaisseaux.
Traitement
Pour ce qui est du traitement des lésions cutanées de dermatomyosite, on l’entreprend uniquement dans les formes amyopathiques primitives, séquellaires ou récidivantes, mais pas dans les formes myopathiques car, dans ces dernières, c’est le traitement de l’atteinte musculaire qui contribuera à la guérison des lésions cutanées. Les traitements topiques comprennent la photoprotection, les dermocorticoïdes et le tacrolimus topique, qui semble être plus efficace que dans les lésions lupiques. Les traitements généraux comprennent les anti-paludéens de synthèse, le mycophénolate mofétil, le méthotrexate, la dapsone et les immunoglobulines intraveineuses.
2. Les lésions vasculaires
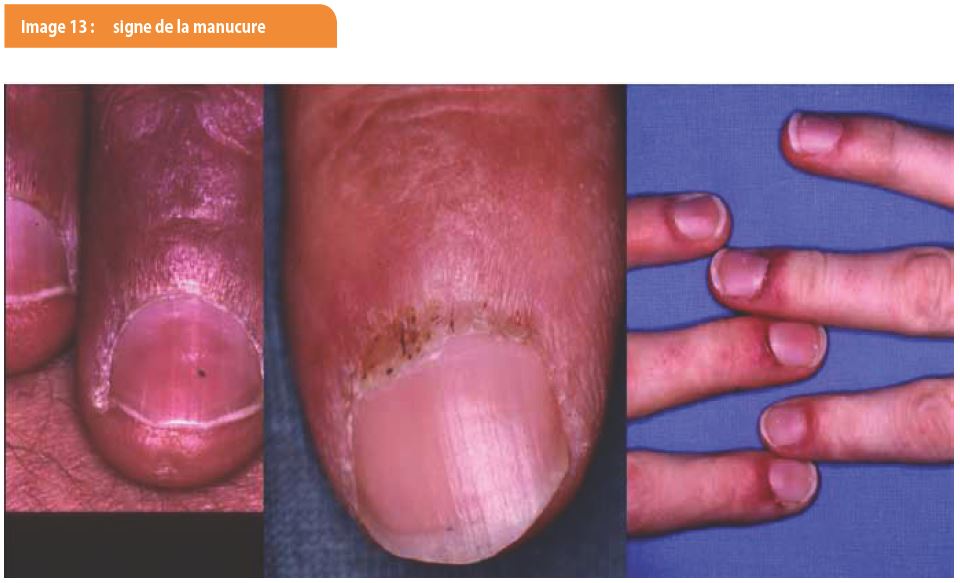
L’érythème congestif de la sertissure des ongles associé à un repli unguéal douloureux à la pression ou au refoulement des cuticules constitue le signe de la manucure (Image 13). Les mégacapillaires sont visibles à la capillaroscopie et parfois même à l’œil nu. On peut également retrouver des vascularites ou des thromboses, surtout dans les formes associées à une néoplasie ou à une autre connectivite.
3. Les lésions non spécifiques non vasculaires
Enfin, les lésions non spécifiques non vasculaires comprennent les panniculites, les « mains mécaniques » du syndrome anti-synthétases, les calcifications cutanées, les mucinoses en plaques, l’œdème associé à la myosite et l’aspect de pityriasis rubra pilaire associé à une kératodermie palmoplantaire, typique de la DM de type Wong.
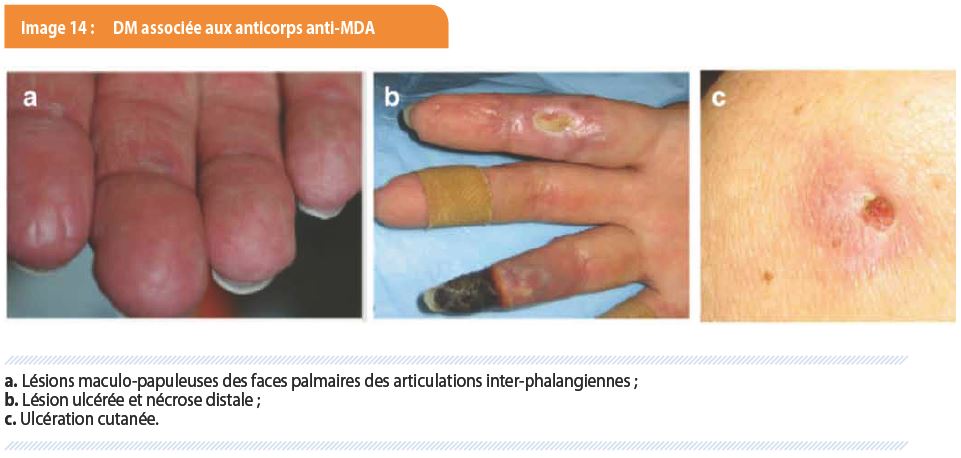
Le Professeur Francès a terminé son exposé en nous présentant une forme sérologique particulière de dermatomyosite, à savoir la DM associée aux anticorps anti-MDA. Celle-ci se caractérise par des lésions maculo-papuleuses localisées aux faces palmaires des articulations métacarpo-phalangiennes ou inter-phalangiennes, avec une surface érythémateuse, hyperkératosique ou ulcérée. On y retrouve également des lésions de panniculite, des ulcérations cutanées fréquentes et des nécroses cutanées (Image 14). L’atteinte musculaire est discrète ou absente et c’est l’atteinte pulmonaire qui conditionne le pronostic.
LES AUTO-ANTICORPS DANS LES RHUMATISMES SYSTÉMIQUES : SIMPLIFIONS CE CASSE-TÊTE !
F. Houssiau
PATHOGÉNICITÉ DES AUTO-ANTICORPS
Le Professeur F. Houssiau rappelle que les auto-anticorps ne sont pas que des biomarqueurs. En effet, ils peuvent être eux-mêmes pathogènes. En voici trois exemples :
1. En cas de lupus érythémateux systémique chez une femme enceinte, les auto-anticorps maternels anti-SSA/Ro passent le placenta et peuvent être responsables d’un lupus érythémateux subaigu néonatal sous forme d’une éruption cutanée transitoire, d’une cytopénie et/ou d’un bloc cardiaque congénital.
2. Les anticorps anti-ADN reconnaissent certaines structures antigéniques au sein de la membrane basale glomérulaire et peuvent de la sorte causer une néphrite lupique.
3. Les anticorps anti-phospholipides sont dirigés contre une protéine sérique circulante, la beta2-glycoprotéine-1 (β2GP1), elle-même capable de se lier aux phospholipides. Ils sont pathogènes par leur potentiel thrombogène. En effet, ils peuvent être responsables de thromboses veineuses, d’accidents vasculaires cérébraux et de fausses couches tardives à répétition. Ces anticorps traversent le placenta et peuvent se fixer sur la β2GP1 présente à la surface du trophoblaste. Ils entrainent ainsi une dysfonction trophoblastique provoquant une souffrance placentaire et fœtale. On peut éviter ces altérations par administration d’héparine qui empêche la liaison de la β2GP1 aux phospholipides du trophoblaste.
QUAND FAUT-IL RECHERCHER LES AUTO-ANTICORPS ?
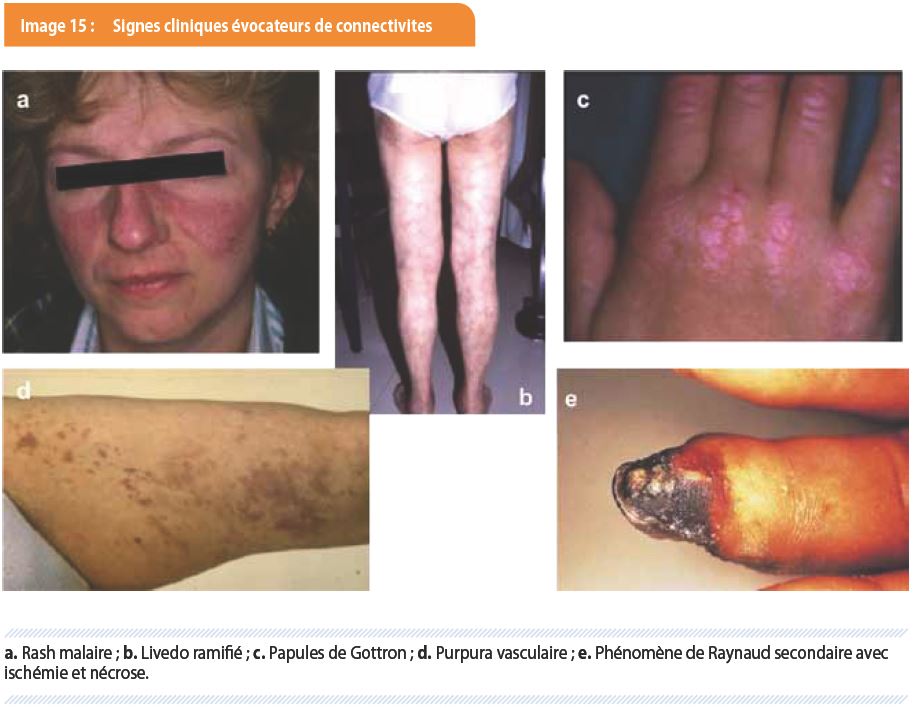
Ou plutôt, quand ne faut-il pas les rechercher : notamment, en cas de contexte clinique peu évocateur, de plaintes vagues et aspécifiques et de syndrome polyalgique idiopathie diffus, communément appelé fibromyalgie. Par contre, en présence de signes cliniques évocateurs, notamment en cas de rash malaire, de livedo ramifié, de papules de Gottron, de purpura vasculaire et de phénomène de Raynaud secondaire, la recherche de ces anticorps est justifiée (Image 15).
La recherche des auto-anticorps est également justifiée en cas d’atteinte systémique, à savoir, de pleurésie, de péricardite, de glomérulonéphrite segmentaire et focale nécrosante, d’alvéolite hémorragique, de fibrose pulmonaire, de myosite ou encore de myélite transverse.
QUELS TESTS FAUT-IL REALISER ?
1. Les anticorps antinucléaires
Il faut commencer par rechercher les anticorps antinucléaires ou facteur antinucléaire (FAN) par technique d’immunofluorescence indirecte. Le titre est une dilution et la technique est donc observateur-dépendant. Le FAN est parfaitement sensible pour le lupus érythémateux disséminé actif mais n’est nullement spécifique. On retrouve des anticorps antinucléaires dans > 95 % des lupus érythémateux systémique, 80 % des syndromes de Sjögren, 80 % des sclérodermies et 40 % des dermatomyosites.
On peut également en retrouver, à des titres faibles, dans des infections chroniques, dans des dysthyroïdies auto-immunes et dans la population générale, surtout chez les personnes âgées. Il est intéressant de rechercher les anticorps antithyroïdiens en cas de FAN faiblement positif, celui-ci pouvant être lié à l’auto-immunité associée à la thyroïdite d’Hashimoto.
On peut donc banaliser la présence d’anticorps antinucléaire en cas d’absence de contexte clinique, de titres faibles et/ou d’absence de spécificité antigénique.
2. Les auto-anticorps spécifiques dans les rhumatismes systémiques
En cas de probabilité pré-test faible, on ne recherche les spécificités antigéniques que si le FAN est positif. Si la probabilité pré-test est forte, on associe d’emblée à la recherche du FAN celle des spécificités antigéniques.
La recherche des anticorps anti-ADN peut se faire par technique de radio-immuno-essai selon Farr (RIA) et par test sur « Crithidia luciliae » mais pas par Elisa car il y a trop de faux positifs. La technique RIA est très quantitative. Celle sur « Crithidia luciliae » est quant à elle très spécifique mais difficile à réaliser et à quantifier.
La recherche des anticorps anti-ENA se fait par Elisa screen (RNP, Sm, SSA, SSAB, CENP-B, Scl70, Jo1), Elisa spécifiques et immunoblot.
Les auto-anticorps associés au lupus érythémateux disséminé sont les suivants :
- les anticorps anti-ADN, excellents marqueurs de la néphrite lupique ;
- les anticorps anti-Sm, plus fréquents chez les malades africains ou afro-américains ;
- les anticorps anti-SSA/SSB, associés au lupus érythémateux subaigu et à la triade classique : atteinte cutanée, atteinte articulaire et syndrome sec ;
- les anticorps anti-RNP, associés à la présence de doigts boudinés, de myosite et à un risque accru d’hypertension artérielle pulmonaire.
Les auto-anticorps spécifiques du syndrome anti-phospholipides (SAPL) sont les anticorps anti-cardiolipines, l’anticoagulant lupique et les anticorps anti-β2-glycoprotéine1 :
- Les anticorps anti-cardiolipines sont détectés par Elisa et leur taux est significatif s’il dépasse 30 UGPL. Ces anticorps peuvent être de type IgM ou IgG mais ces derniers sont plus fréquemment associés aux symptômes cliniques (thromboses). -
- L’anticoagulant lupique est détecté par le test d’anticoagulant du lupus ou antiprothrombinase, qui est un test de coagulation phospholipides-dépendant. L’anticoagulant lupique est responsable d’un allongement du temps de céphaline activée (TCA) lors des tests de laboratoire et le diagnostic différentiel se pose donc avec certains troubles de la coagulation (hémophilie, von Willebrand). On peut doser l’anticoagulant du lupus chez un patient sous anti-vitamine K pour autant que l’INR ne soit pas supérieur à 3. Par contre, si le patient est sous héparine, même à dose prophylactique, le test n’a aucune valeur et ne doit pas être demandé.
- Les anticorps anti-β2-glycoprotéine1, détectés par Elisa, sont dirigés contre cette protéine et non contre les phospholipides eux-mêmes.
Les auto-anticorps des myosites comprennent :
- D’une part, les anticorps spécifiques des myosites :
- Les anticorps anti-synthétases, dont l’anticorps anti-Jo1 est le plus connu. Le tableau clinique du syndrome anti-synthétase comprend une fièvre, une myosite, un phénomène de Raynaud, une arthropathie inflammatoire, une fibrose pulmonaire, des « mains mécaniques » et parfois un rash.
- Les anticorps associés aux myopathies nécrosantes : anticorps anti-SRP et anti-HMGCR. Ces myopathies nécrosantes peuvent être induites par des statines et sont associées à des taux extrêmement élevés de CPK. Les anticorps associés à la dermatomyosite amyopathique : anticorps anti-SAE et anti-MDA-5.
- Les anticorps associés à la dermatomyosite : anticorps anti-NXP-2, anti-TIF1γ et anti-Mi-2. L’anticorps anti-TIF1γ est associé à des formes paranéoplasiques de DM.
- D’autre part, les anticorps associés aux myosites mais non spécifiques de celles-ci : anti-SSA/RO, anti-SSB/La, anti-U1-RNP, anti-U3-RNP, anti PM-Scl, anti-Ku. En effet, on peut retrouver ces anticorps dans des formes de chevauchement avec le lupus érythémateux systémique, la sclérodermie systémique, le syndrome de Sjögren, la connectivite mixte, la polyarthrite rhumatoïde et le syndrome des anticorps anti-phospholipides.
Parmi les auto-anticorps de la sclérose systémique, on recherche :
- D’une part, les anticorps classiques de la sclérose systémique :
- Les anticorps anti-topoisomérase 1, également appelés anti-Scl 70, associés à la sclérose systémique diffuse.
- Les anticorps anti-centromère (CENP-B), associés à la sclérose systémique limitée, anciennement appelé syndrome de CREST.
- D’autre part, d’autres anticorps :
- Les anticorps anti-Th/To dans les formes limitées, associées à de l’hypertension artérielle pulmonaire.
- Les anticorps anti-RNA polymérase III dans les formes diffuses, associées à des crises rénales.
- Les anticorps anti-PM-Scl dans les formes de chevauchement avec la myosite.
- Les anticorps anti-U3-RNP dans les formes diffuses, chez les hommes africains, de mauvais pronostic.
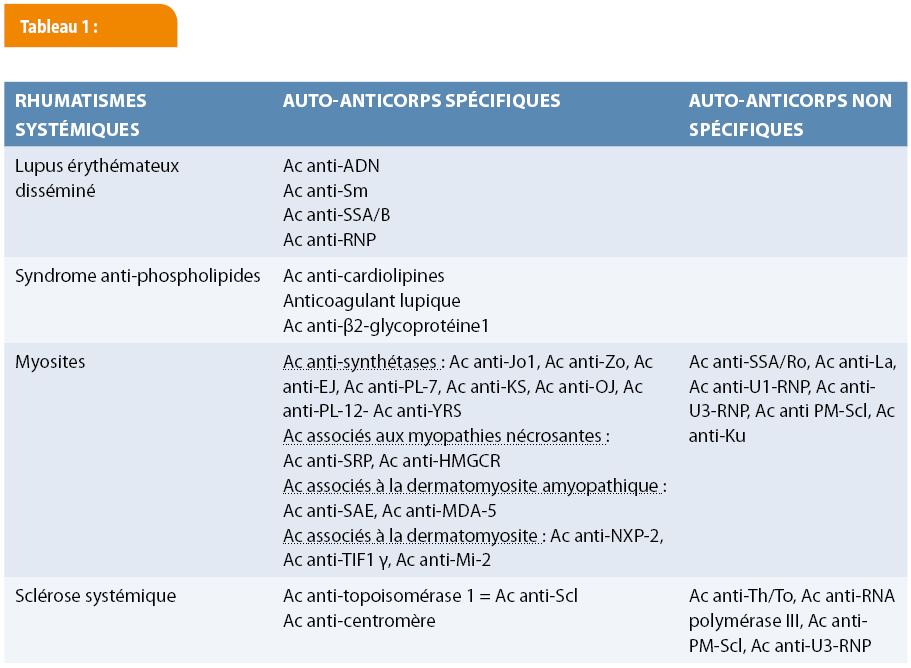
Le tableau 1 reprend les différents auto-anticorps associés aux rhumatismes systémiques évoqués ci-dessus.
Les anticorps anti-cytoplasme des neutrophiles (ANCA) sont des anticorps dirigés contre les antigènes du cytoplasme des granulocytes neutrophiles. Leur dépistage s’effectue en immunofluorescence indirecte (IFI) sur des frottis de polynucléaires neutrophiles et permet de définir deux types d’anticorps en fonction de la localisation de la fluorescence : cytoplasmique pour les c-ANCA et périnucléaire pour les p-ANCA.
Les ANCA sont classiquement retrouvés au cours des vascularites systémiques nécrosantes touchant les vaisseaux de moyen et petit calibre. Mais, ils ne sont pas spécifiques des vasculites. On les retrouve notamment dans 60 % des rectocolites ulcéro-hémorragiques et 20 % des maladies de Crohn. Certains médicaments peuvent induire des ANCA, à savoir le propylthiouracile, la minocycline, l’hydréa et même le lévamisole (produit de coupe de la cocaïne). Enfin, on peut en rencontrer dans la mucoviscidose et dans toutes les maladies inflammatoires chroniques.
Selon la probabilité pré-test, on recherche les spécificités antigéniques anti-protéinase 3 (anti-PR3) et anti-myéloperoxydase (anti-MPO) d’emblée ou uniquement en cas d’ANCA positifs. Dans les vascularites systémiques nécrosantes, les ANCA sont d’excellents marqueurs diagnostiques et le type de sérologie peut orienter le diagnostic vers la classe de la maladie. En effet, l’aspect c-ANCA en IFI et la spécificité anti-PR3 en Elisa sont caractéristiques de la granulomatose avec polyangéite de Wegener. L’aspect p-ANCA en IFI et la spécificité anti-MPO en Elisa sont quant à eux des marqueurs de la polyangéite microscopique, de la granulomatose éosinophilique avec polyangéite (exemple : syndrome de Churg et Strauss) et des glomérulonéphrites nécrosantes focales.
LES TAUX SONT-ILS IMPORTANTS ?
Oui car l’auto-immunité est physiologique. Des titres faibles sont très fréquents avec l’âge, dans les maladies inflammatoires chroniques et en cas de dysthyroïdies. De plus, les auto-anticorps pathogènes ont une haute affinité pour l’antigène (mutations somatiques) et sont présents en (très) grande quantité. Dès lors, seuls des titres élevés doivent être pris en considération dans un contexte clinique cohérent.
FAUT-IL RÉPÉTER LES DOSAGES ?
Non, sauf dans deux circonstances :
1. Pour confirmer la positivité avec des techniques appropriées ;
2. Certains tests sont très quantitatifs et leurs variations corrèlent avec l’activité clinique de la maladie, voire prédisent les poussées cliniques. C’est le cas pour les anticorps anti-ADN, anti-PR3 et anti-MPO. En effet, la récidive de lupus est souvent prédite par l’élévation des anticorps anti-ADN et la chute du complément qui la précède de quelques mois. Dès lors, chez les patients atteints de lupus systémique avec anticorps anti-ADN positifs, il faut les tester à chaque consultation. Il en va probablement de même pour les anticorps anti-PR3 et anti-MPO dans les vasculites systémiques nécrosantes. Par contre, les dosages de tous les autres anticorps ne doivent pas être répétés.
QUAND APPARAISSENT LES AUTO-ANTICORPS ?
L’apparition des auto-anticorps précède les manifestations des maladies auto-immunes. Ceci amène la question du bénéfice potentiel du dépistage de ces auto-anticorps. Cependant, il est inutile de les demander si la clinique ne corrobore pas l’information que ces tests pourraient nous donner.
TAKE HOME MESSAGES
Pour conclure, quatre messages principaux sont à retenir : Il ne faut jamais demander de tests sérologiques rhumatismaux si le contexte clinique n’est pas relevant. Il faut banaliser les titres faibles. Sauf exception, il ne faut pas répéter les dosages. Demain, la classification des syndromes systémiques ne sera sans doute plus clinique mais sérologique et moléculaire.