Introduction
La sclérose tubéreuse de Bourneville est une maladie multi-systémique rare, de transmission autosomique dominante, due à une mutation sur le gène TSC1, codant l’hamartine, ou sur le gène TSC2, codant la tubérine. Deux-tiers des cas sont sporadiques (1).
Elle se caractérise par le développement d’hamartomes dans de nombreux organes dont les principaux sont la peau, le cerveau, les reins, les yeux, le cœur et les poumons (2). Les manifestations cliniques sont très variables avec une expressivité différente (3).
Cette maladie a une pénétrance complète. À l’âge adulte, les patients présentent donc au moins une atteinte évocatrice cérébrale, cutanéo-muqueuse, rénale, rétinienne ou pulmonaire (4).
Les modes de révélation les plus fréquents sont neurologiques, cutanés et cardiaques, et la triade classique d’épilepsie, de retard mental et d’angiofibromes cutanés (5) se retrouve majoritairement chez l’enfant (4).
Les atteintes pulmonaires sont rares et surviennent essentiellement chez la femme adulte sous forme de pneumothorax récidivants ou d’insuffisance respiratoire chronique, secondaires à des lésions de lymphangiomyomatose pulmonaire (6).
L’espérance de vie est majoritairement normale. Néanmoins, la gravité de la maladie est surtout liée aux lésions cérébrales (épilepsie, retard mental et parfois aussi hydrocéphalie) et aux lésions viscérales (complications tumorales rénales, avec risque d’hémorragie ou atteinte de la fonction rénale et pulmonaire) qui peuvent mettre en jeu le pronostic vital (7).
Vignette clinique
Une patiente de 25 ans sans aucuns antécédents médicaux-chirugicaux ou familliaux, se présente aux urgences pour un pneumothorax diagnostiqué sur une radiographie thoracique. (Figure 1). Celle-ci fume 15 cigarettes par jour (10 UAP) et prend une contraception orale.
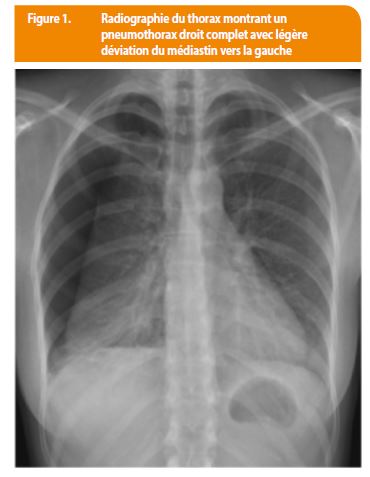
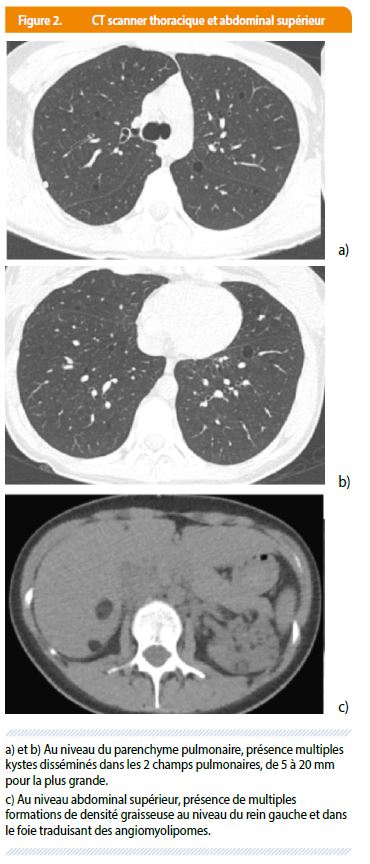
Trois semaines plus tard, la patiente récidive son pneumothorax droit. Lors du bilan le scanner thoracique montre des lésions kystiques à parois très fines distribuées aléatoirement dans les deux poumons. Sur les coupes inférieures passant par l’abdomen supérieur on objective des lésions de densité graisseuse dans le parenchyme du rein gauche et du foie, lésions évocatrices d’angiomyolipomes (Figure 2). L’association de kystes pulmonaires et d’angiomyolipomes abdominaux sont très évocateurs de LAM, voire de STB.
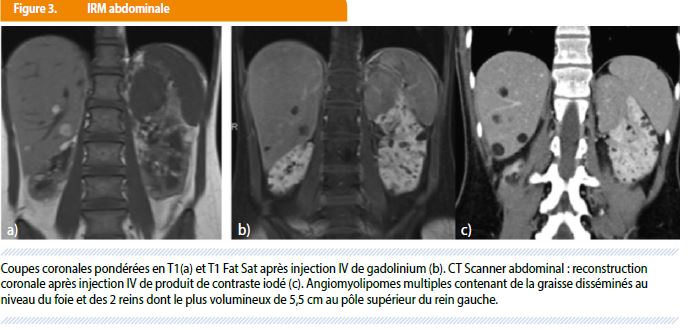
Un bilan d’extension est dès lors réalisé pour confirmer les hamartomes abdominaux et rechercher d’autres signes de STB : le scanner abdominal et l’IRM hépato-rénale mettent en évidence une infiltration endo-hépatique et surtout rénale, bilatérale, par de multiples lésions à caractère partiellement graisseux correspondant à des angiomyolipomes dont le plus volumineux mesure 5,5 cm au pôle supérieur du rein gauche (Figure 3).
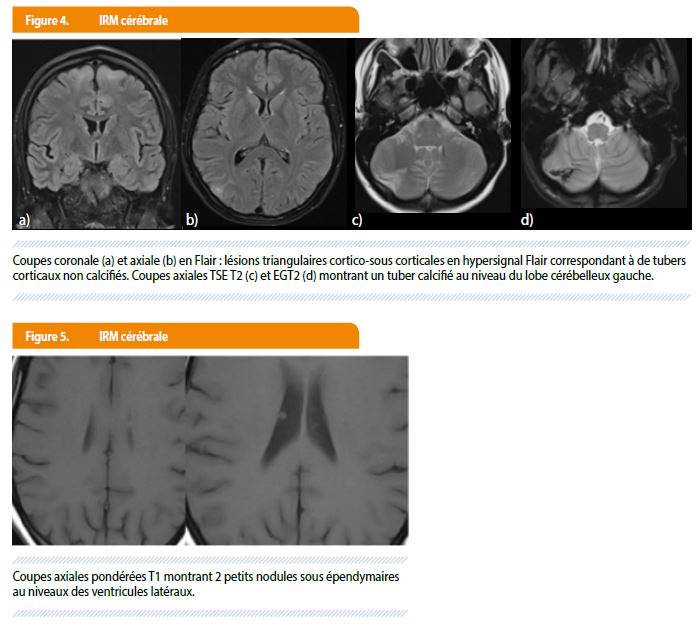
L’IRM cérébrale retrouve des anomalies typiques de STB avec plusieurs tubers corticaux sus et sous tentoriels (Figure 4) associés à des nodules sous épendymaires au niveau des ventricules latéraux (Figure 5).
Les examens ophtalmologique et dermatologique sont tout à fait normaux. À la suite de ce bilan, l’équipe médicale a conclu à une récidive de pneumothorax droit sur lymphangioleiomyomatose pulmonaire développée dans le cadre d’une sclérose tubéreuse de Bourneville.
Une semaine après la sortie de son hospitalisation, la patiente est revenue pour une nouvelle récidive de pneumothorax droit ayant mené à un talcage au cours duquel une biopsie pulmonaire a été réalisée confirmant d’un point de vue histologique des lésions de lymphangioléiomyomatose.
La suite du bilan de sclérose tubéreuse de Bourneville a été poursuivi en milieu universitaire. Les spécialistes confirment l’absence totale de manifestation de STB avant son premier pneumothorax notamment d’un point de vue neurologique et cutané. Elle a bénéficié d’une échographie cardiaque normale et de tests fonctionnels respiratoires également normaux.
Les médecins du service de médecine interne lui ont conseillé l’arrêt des oestroprogestatifs et de poursuivre avec une contraception non hormonale. Le dossier a été discuté, en réunion pluridisciplinaire afin d’évaluer l’opportunité d’un traitement par mTORi. La patiente présetant un angiomyolipome rénal de 5,5cm un traitement par everolimus a éte proposé en vue de ralentir l’évolution de la maladie. Elle a été adressée à la consultation multidisciplinaire consacrée à la sclérose tubéreuse de Bourneville à l’institut des maladies rares des cliniques universitaires Saint-Luc et un conseil génétique a été proposé à la patiente. Le bilan génétique de cette jeune femme montre une forme sporadique de STB, la patiente est identifiée porteuse d’une duplication intra-génique dans le gène TSC2. Il n’existe aucuns antécédents familiaux de STB.
Discussion
Diagnostic de LAM et de STB
Diagnostic de LAM
La lymphangiommyomatose est une maladie pulmonaire rare caractérisée par une prolifération de cellules de type musculaire lisse qui entraîne la formation de kystes multiples pulmonaires.(9)
Elle survient :
- soit de manière sporadique ( environ 1/400 000 femmes adultes) ;
- soit dans le cadre de la STB où elle affecte 30 à 40 % des femmes adultes ( 9).
La LAM associée à la STB doit être différentiée de la LAM dite sporadique qui se caractérise uniquement par des lésions pulmonaires et fréquemment des angiomyolipomes rénaux, sans les autres manifestations de STB. Par ailleurs, la LAM sporadique ne présente pas de risque de transmission génétique, les anomalies génétiques étant essentiellement somatiques sans mutation germinale. Dans le LAM sporadique, l’atteinte pulmonaire est significativement plus diffuse alors que dans la LAM associée à la STB, on observe plus d’angiomyolipomes hépatiques et rénaux. Néanmoins, on retrouve des points communs entre la LAM sporadique et la LAM associée d’une STB comme le terrain féminin, le tableau clinique respiratoire identique et la grande fréquence d’angiomyolipomes rénaux. Malgré leurs points communs, ces deux pathologies représentent deux entités différentes qu’il importe de différencier (8).
Le diagnostic de certitude de la LAM est déterminé sur une biopsie pulmonaire et/ou l’association d’un tableau clinique et d’un aspect de scanner thoracique caractéristique (9).
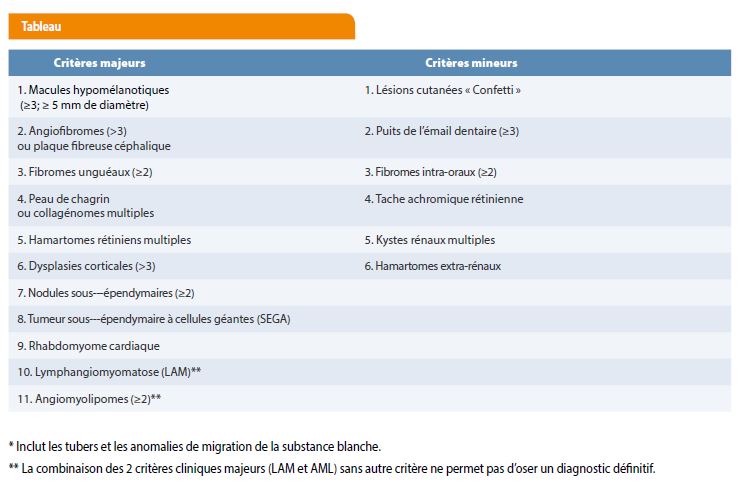
Des critères diagnostiques de LAM chez la femme ont été proposés par la European Respiratory Society repris dans le tableau ci-dessous (9-10).

Afin de diagnostiquer une LAM, il est nécessaire d’exclure d’autres causes de maladie kystique pulmonaire comme l’histyocytose langerhansienne, la pneumopathie interstitielle lymphocytaire, l’emphysème centro-lobulaire, … Un bilan diagnostic complet pour ces autres causes est indispensable lors de l’évaluation initiale chez les patientes avec LAM probable et LAM possible (9).
Diagnostic de STB
La sclérose tubéreuse de Bourneville est diagnostiquée sur base de critères consensuels qui sont des critères diagnostiques cliniques et un critère diagnostique génétique (3).
Les critères diagnostiques cliniques, combinant un examen physique et des tests d’imagerie, ont été établis par M. Gomez en 1988 et ceux-ci ont été revus lors de la conférence internationale de consensus sur la sclérose tubéreuse de Bourneville de 2012 à Washington. Selon la combinaison de ces critères, il est déterminé si le diagnostic est certain ou possible (6).
Diagnostic certain de la STB:
- 2 critères cliniques majeurs ou 1 critère majeur + au moins 2 critères mineurs
ou
- Critère génétique indépendant.
Diagnostic possible de la STB :
- 1 critère majeur ou au moins 2 critères mineurs.
Le critère diagnostique génétique consistant en l’identification d’une mutation reconnue comme pathogène dans un des 2 gènes TSC1 ou TSC2 suffit à établir le diagnostic de la STB. La mutation d’un des gènes responsables n’est identifiée que chez 85 % des personnes atteintes. Un résultat négatif n’exclut pas le diagnostic, et il faudra recourir aux critères cliniques ci-dessus (3).
La cause génétique de la STB chez le 15% des patients chez qui une mutation n’est pas trouvée est jusqu’à ce jour inconnue. Ceux-ci ont généralement une maladie clinique plus douce que les patients atteints de mutations TSC1 ou TSC2 identifiées (11). Statistiquement, une mutation située sur le gène TSC2 est associée à une maladie plus sévère qu’une mutation TSC1 mais il existe de nombreuses exceptions individuelles (11). Les patients atteints d’une mutation TSC2 ont été diagnostiqués, en moyenne, 9 ans avant ceux atteints de mutations TSC1 et 11 ans avant ceux sans mutations identifiables (12).
Il est toutefois impossible de prédire la gravité de la maladie en fonction de la mutation retrouvée, car il existe une grande variabilité, même chez deux patients affectés par la même mutation (7-11-13). Les mutations TSC2 sont plus fréquentes que les mutations TSC1 retrouvées seulement dans 10 à% à 30% des familles. Dans la STB sporadique, il existe un excès encore plus important de mutations TSC2 (13).
Traitement et surveillance
Il n’existe pas à l’heure actuelle de traitement permettant de guérir la sclérose tubéreuse de Bourneville. Jusqu’il y a peu les possibilités de traitement dans les atteintes sévères se limitaient à des traitements symptomatiques (antiépileptiques, drainage des pneumothorax, embolisation des angiomyolipomes hémorragiques) ou ablatifs. Ces dernières années, les inhibiteurs mTORi, premières molécules ayant une action ciblée et multi-systémique, ont révolutionné le traitement de la maladie. Ce sont les premières molécules permettant d’infléchir le cours de la maladie et d’éviter éventuellement le recours à la chirurgie (3). Ces traitements mTORi ont fait l’objet de recommandations internationales publiées en 2013 (14).
Les indications actuelles du traitement par mTORi sont les suivantes
- Astrocytome cérébral asymptomatique de volume croissant et potentiellement menaçant (14) ou lorsque la chirurgie comporte un risque ou n’est pas possible (6).
-Atteinte pulmonaire réduisant la fonction respiratoire (14).
-AML rénaux en croissance ou à risque d’hémorragie (diamètre de plus de 3 cm) et/ou ayant déjà conduit à une réduction du capital néphrotique. L’embolisation sélective, voire une néphrectomie partielle, est aujourd’hui réservée au traitement d’urgence d’un AML hémorragique. Ces traitements amputent clairement la fonction des reins et peuvent même aboutir à une insuffisance terminale (14). Par contre, les kystes rénaux ne nécessitent aucun traitement (3-7).
Les mTORi sont capables de stabiliser voire même de réduire la taille des hamartomes dans tous les tissus atteints (14). La STB peut donc être prise en charge de manière causale sur la base des connaissances de la biologie moléculaire (2). Par contre, l’arrêt du traitement mTORi est inévitablement suivi d’une réaugmentation du volume des hamartomes. Cela implique qu’ils devront être pris sans interruption pour conserver leur efficacité (14). Les effets secondaires du traitement mTORi sont acceptables au moyen de stratégie de dosage prudente parce que ceux-ci ont une fenêtre thérapeutique étroite (15).
Les effets secondaires les plus fréquents sont les stomatites, l’acné, les diarrhées, les céphalées, les dyslipidémies, l’œdème des membres inférieurs, l’albuminurie, la eucopénie, l’infection respiratoire, la formation de kystes ovariens et l’apparition de trouble du cycle menstruel mais ils ont tendance à s’atténuer avec le temps (2-11-15).
L’arrêt des oestroprogestatifs est préconisé puisqu’on pense que les œstrogènes sont susceptibles de favoriser le développement des lésions rénales et pulmonaires. Bien que cela ne soit pas formellement établi, on conseille d’éviter ce mode de contraception mais aussi le traitement hormonal substitutif de la ménopause. Toutefois, un moyen de contraception efficace devra être employé par toutes les femmes qui seront traitées par mTORi puisque celui-ci a un effet tératogène.
Également, la grossesse a été longtemps déconseillée car on a observé des aggravations de la LAM à cette occasion. Néanmoins, le rôle de la grossesse dans l’aggravation de la maladie n’est pas prouvé. Une évaluation approfondie au cas par cas est donc nécessaire.
Par ailleurs, un soutien psychologique est souhaitable car c’est une maladie chronique qui peut être très invalidante (9).
Pour la surveillance en cas de STB , il est recommandé
- Sur le plan neurologique : d’effectuer une IRM tous les 1 à 3 ans généralement jusqu’à l’âge de 25 ans. À l’âge adulte seuls les individus porteurs d’astrocytomes subépendymaire à cellules géante seront suivis à intervalle régulier compte tenu du potentiel de croissance de ce type de tumeur (6).
- Sur le plan pulmonaire : dès qu’une LAM est diagnostiquée de réaliser tous les 2 à 3 ans un scanner thoracique. Il est également conseillé de faire une évaluation de la fonction pulmonaire avec un test de marche de 6 minutes et un EFR annuellement (6).
- Sur le plan rénal : de réaliser une échographique rénal, un scanner abdominal ou IRM si possible tous les 1 à 3 ans dès le diagnostic de la STB et de vérifier une fois par an la fonction rénale et la tension artérielle. Dès qu’on découvre un angiomyolipome rénal, une imagerie annuelle est recommandée (6-8).
- Sur le plan cardiaque : De réaliser un ECG tous les 3 à 5 ans pour détecter d’éventuels défauts de conduction chez des patients asymptomatiques de tous âges (6).
- Sur le plan dermatologique et ophtalmologique, un examen clinique annuel est recommandé (6).
- Cette surveillance ciblée, par des examens de contrôle réguliers, permet de prévenir l’apparition de complications cérébrales, rénales et pulmonaires pouvant mettre en jeu le pronostic vital (4).
De plus, il recommandé de proposer des tests génétiques en consultation spécialisée avec des conseils génétiques chez les personnes en âge de procréer (6) et de référer les patients dans les centres experts de la STB, dans lesquels une consultation multidisciplinaire va permettre de coordonner les avis et de générer un compte rendu unique (14).
De même, il est important de recueillir tous les cas de STB dans une banque de données qui permet de mieux comprendre les manifestations de la STB et facilite le développement de meilleures stratégies de gestion et de surveillance pour les patients atteints de TSC (16).
Conclusion
La STB est une maladie génétique rare, de transmission dominante, avec une grande variété de symptômes, surtout neurologiques et cutanés chez l’enfant.
Dans 20 % des cas elle se manifeste tardivement chez l’adulte avec un phénotype différent; les lésions cutanées sont moins fréquentes, les symptômes sont essentiellement liés aux lésions rénales et chez la femme aux lésions pulmonaires. L’histoire familiale est absente dans 2/3 des cas.
En cas de pneumothorax spontané récidivant chez une femme adulte, il faut donc rester vigilant et garder à l'esprit la possibilité du diagnostic de LAM dans le cadre d’une STB.
Devant toute lésion kystique pulmonaire, une recherche de STB est préconisé même s’il n’existe aucune histoire familiale.
Il faut savoir différencier la LAM sporadique de la LAM associée à une STB.
Il est recommandé de diagnostiquer la STB sur base de critères consensuels qui sont un critère diagnostique génétique et des critères diagnostiques cliniques. Il est primordial de référer les patients à un centre d’expert avec une approche pluridisciplinaire afin d'optimaliser la prise en charge globale pour les patients atteints de STB.
Affiliations
(1) Service de radiologie médicale au CHwapi de Tournai- Site Union
Correspondance
Dr. Céline Brasseur celine.brasseur@student.uclouvain.be
Références
- Henske EP, Jozwiak S, Kingswood JC, Sampson JR,Thiele E. A.Tuberous sclerosis complex, Nature Reviews Disease Primers 26 May 20162:16035. doi: 10.1038/nrdp.2016.35.
ouvrir dans Pubmed - Serra A, Bonny O, Burki S, Dorn T , Fuster D, Guzman R, et al. La sclérose tubéreuse de Bourneville: pathogenèse, clinique et nouvelles options thérapeutiques. Forum Med Suisse. 2013; 13 (36) : 696–702.
- Dekeuleneer V, Ho T-A, Van Rijckevorsel, Sznajer Y, Nassogne M-C, Godefroid N., et al. Les inhibiteurs mTOR : nouvel outil thérapeutique dans la prise en charge de la sclérose tubéreuse de Bourneville. Louvain Med. 2015; 134 (10): 537-546.
- Chipaux M, Chiron C, Touraine R, Ouss L, Dulac O, Nabbout R. Sclérose tubéreuse de Bourneville : mise au point et actualités. Épilepsies. 2009 ; 21 (1) : 34-40.
- Seibert D, Hong CH, Takeuchi F, Olsen C, Hathaway O, Moss J, et al. Recognition of tuberous sclerosis in adult women: delayed presentation with life- threatening consequences. Ann Intern Med. 2011 Jun 21;154(12):806-13, W-294
ouvrir dans Pubmed - Krueger DA, Northrup H. Tuberous Sclerosis Complex Surveillance and Management: Recommendations of the 2012 International Tuberous Sclerosis. Complex Consensus Conference Pediatric Neurology. 49 (2013) : 255-265.
ouvrir dans Pubmed - Chiron C. Sclérose tubéreuse de Bourneville: spectres cliniques, démarche diagnostique, prise en charge pratique. La Lettre du Neurologue - Vol. XII - n° 1-2 - janvier-février 2008.
- Nassaf M, Afif H, Aichane A, Ridai M, Souabny A, Ouafi NE, et al. Sclérose tubéreuse de Bourneville révélée par des pneumothorax à répétition. La Revue de médecine interne. 2008 ; 29 : 252-254.
ouvrir dans Pubmed. - Lymphangioléiomyomatose: Protocole national de diagnostic et de soins pour les maladies rares, HAS / Service des bonnes pratiques professionnelles / Mars 2012, site disponible sur : https://www.has-sante.fr/portail/upload/docs/application/pdf/2012-10/ald...
- Johnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, Harari S, et al. European respiratory society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010; 35: 14–26.
ouvrir dans Pubmed - Crino P, Nathanson KL, Henske EP. The Tuberous Sclerosis Complex. N Engl J Med. 2006; 355: 1345-56.
ouvrir dans Pubmed - Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. PEDIATRICS. 2011; 127 (1): e117-25. doi: 10.1542/peds.2010-0192.
ouvrir dans Pubmed - Rosset C, Brinckmann Oliveira Netto C, Ashton-Proll P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genetics and Molecular Biology. 2017; 40 (1): 69-79.
ouvrir dans Pubmed - Pirson Y, HO T-A, Demoulin N, Godefroid N, Dekeuleneer V, van Rijckevorsel K, et al. Sclérose tubéreuse de Bourneville: poser le diagnostic et traiter à bon escient, Louvain Med. 2017 ; 136 (1) : 10-15.
- Urban T, Lymphangioleiomyomatose pulmonaire avec ou sans sclérose tubéreuse de Bourneville. Rev Mal Respir. 2007; 24 : 725-40.
ouvrir dans Pubmed - Kingswood JC, D’Augeres GB, Belousova E, Ferreira JC, Carter T, Castellana R, et al. Tuberous sclerosis registry to increase disease awareness (Tosca)- baseline data on 2093 patients, Orphanet Journal of Rare Diseases. 2017; 12:2.
ouvrir dans Pubmed