Introduction
La maladie de Vogt Koyanagi Harada (VKH) est une maladie auto-immune multi-systémique dont la présentation clinique implique d’ordinaire l’ophtalmologue en première ligne (1,2). Le cas rapporté illustre le défi que représente la prise en charge de cette affection oculaire qui menace le pronostic visuel du patient.
Observation
Un jeune homme d’origine marocaine, âgé de 17 ans, est référé en urgence pour baisse d’acuité visuelle bilatérale accompagnée de céphalées et otalgies, la fluoangiographie (FA) réalisée par l’ophtalmologue référent révèle des décollements rétiniens exsudatifs aux deux yeux (Figure 1). Il ne rapporte aucun antécédent médical et ne prend aucun médicament.
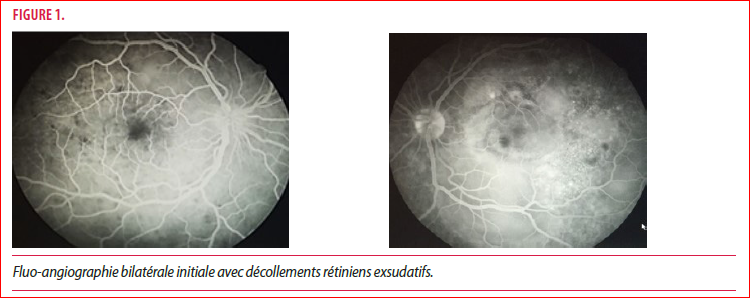
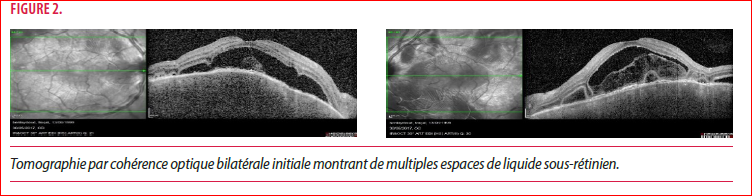
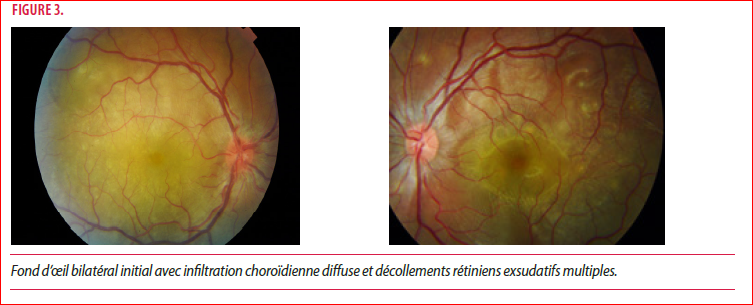
L’acuité visuelle est mesurée à 1/10 de loin et inférieure à 1/10 de près. L’examen à la lampe à fente montre une panuvéite granulomateuse bilatérale avec des décollements rétiniens exsudatifs bien visibles à la tomographie par cohérence optique (OCT) (Figure 2) et un aspect infiltré jaunâtre de la choroïde au pôle postérieur (Figure 3).
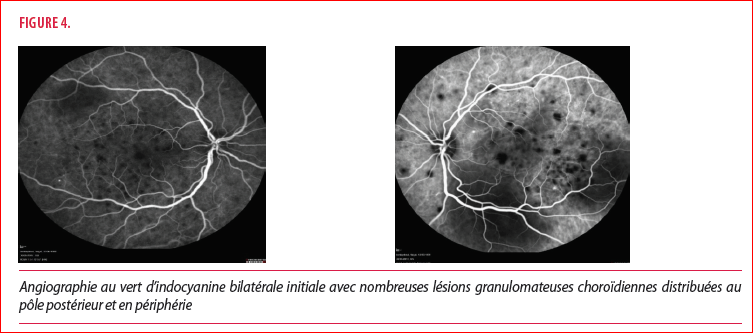
L’angiographie de la choroïde au vert d’indocyanine permet de visualiser de nombreuses lésions granulomateuses choroïdiennes distribuées au pôle postérieur et en périphérie (Figure 4).
L’examen clinique général ainsi que les tests de biologie médicale réalisés par nos confrères internistes se révèlent normaux.
Devant l’aspect clinique très évocateur, le diagnostic de maladie de VKH est retenu et le traitement instauré comprend l’instillation oculaire de prednisolone (PredForte®) à raison d’une goutte par heure et un bolus de methylprednisolone iv 1g/jour durant trois jours suivi d’un relai oral par methylprednisolone 1mg/kg/jour à dose dégressive. L’azathioprine à la dose de 150mg/j est introduite après avoir contrôlé que le patient ne présentait pas de mutation de la S-thiopurine méthyl transférase.
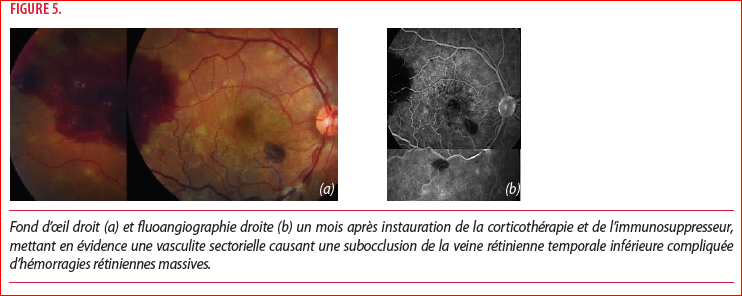
Un mois après l’instauration du traitement, notre patient présente une acuité visuelle normalisée à 10/10 bilatéralement, l’inflammation intraoculaire a quasiment disparu et les décollements rétiniens sont réappliqués. Le patient se plaint cependant de trouble visuel transitoire à l’œil droit et l’examen du fond d’œil (Figure 5) ainsi que la FA (Figure 6) mettent en évidence une image de vasculite sectorielle causant une subocclusion de la veine rétinienne temporale inférieure compliquée d’hémorragies rétiniennes massives. Face à cette présentation inattendue et inexpliquée dans le cadre d’un VKH, un bilan complémentaire est initié et le traitement par trois injections intra vitréennes d’anti-VEGF permet une disparition progressive des hémorragies. Les tests biologiques de coagulation sanguine ainsi que les sérologies HIV et syphilis reviennent négatifs. Le QuantiFERON-TB quant à lui est positif et sera complété par un Pet-scan montrant des ganglions médiastinaux hyper métaboliques. Une biopsie ganglionnaire, par écho endoscopie transbronchique, met en évidence une granulomatose nécrosante et l’analyse par PCR confirme une infection active à mycobacterium tuberculosis.
Devant le diagnostic de réactivation tuberculeuse sous traitement immunosuppresseur, un traitement anti tuberculeux est instauré à base de rifampicine, isoniazide, pyrazinamide, ethambutol et pyridoxine. La dose d’azathioprine est quant à elle réduite à 125mg/jour pour diminuer le risque d’hépatotoxicité secondaire à la combinaison des traitements.
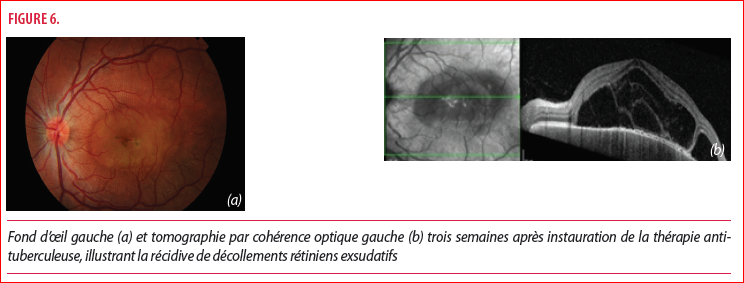
Trois semaines plus tard, le patient se présente avec une baisse d’acuité visuelle brutale de l’œil gauche mesurée à 1.6/10 de loin et 3/10 de près, expliquée par une récidive inflammatoire choroïdienne et la réapparition de décollements rétiniens exsudatifs (Figure 7). La récidive inflammatoire, survenant malgré un traitement immunosuppresseur bien conduit, s’explique par l’induction enzymatique du cytochrome p450 induite par la rifampicine. Cette induction enzymatique entraine une clairance plasmatique de la methylprednisolone d’environ 45% conduisant à la baisse d’efficacité du traitement cortisoné (3,4). Devant cette récidive inflammatoire, le traitement antituberculeux est poursuivi tel quel et le patient reçoit trois bolus de methylprednisolone 750mg/jour avec relais oral à dose très lentement dégressive cette fois.
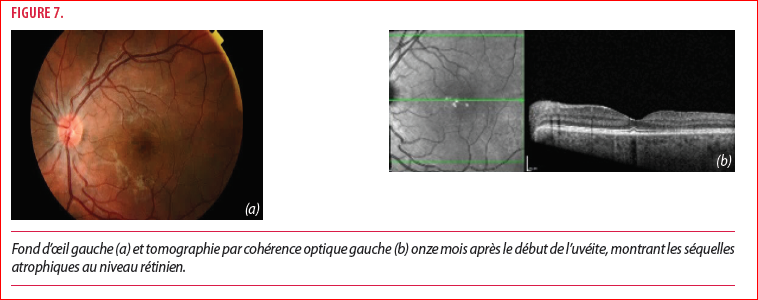
Lors du contrôle à onze mois du début de l’uvéite, le traitement consiste en methylprednisolone 24mg/jour, azathioprine 125mg/jour et le traitement antituberculeux instauré sept mois auparavant. L’acuité visuelle est de 10/10 bilatéralement de loin comme de près et le patient ne présente plus aucun symptôme. L’examen ophtalmologique ne montre aucune activité inflammatoire mais la présence de séquelle atrophique des lésions choroïdiennes et de l’occlusion de la branche veineuse rétinienne.
Discussion
La maladie de Vogt Koyanagi Harada est une pathologie auto-immune multi systémique d’étiologie encore indéterminée. Elle se caractérise par une inflammation de tissus contenant des mélanocytes : méninges, oreille interne, uvée et peau. Il est rapporté une association avec les antigènes HLA DR1 et HLA DR4. Les individus à pigmentation plus marquée, comme notre patient, sont plus susceptibles d’en être affectés (5).
L’histoire naturelle de la maladie se caractérise par quatre phases (6).
- La phase prodromale avec des manifestations neurologiques et auditives qui durent quelques jours : méningite, vertiges, acouphènes, surdité.
- La phase uvéitique qui dure plusieurs semaines et se caractérise par une uvéite granulomateuse bilatérale, une infiltration choroïdienne diffuse, une vitrite, une papillite, des nodules de Dalen Fuchs et des décollements rétiniens exsudatifs.
- La phase chronique qui se caractérise au niveau tégumentaire par une alopécie, un vitiligo et une poliose. Au niveau ophtalmologique, le fond d’œil prend aussi un aspect dépigmenté en « coucher de soleil ».
- Enfin, la phase de récurrence montre des épisodes d’exacerbations d’uvéite antérieure. À ce stade une récidive de décollement de rétine exsudatif ou d’infiltration choroïdienne est rare.
Les critères diagnostiques révisés (7) sont :
- absence de traumatisme oculaire pénétrant ;
- absence d’autre pathologie oculaire ;
- uvéite bilatérale ;
- manifestations auditives ou neurologiques ;
- manifestations tégumentaires.
On parle de VKH oculaire isolé lorsque les trois premiers critères sont présents, de VKH incomplet lorsque s’y ajoute le quatrième ou cinquième critère et enfin de VKH complet lorsque les cinq critères sont retrouvés. Notre patient présentait donc un VKH incomplet puisque les manifestations tégumentaires n’étaient pas apparues avant l’instauration du traitement.
Le traitement du VKH repose sur la corticothérapie à haute dose débutant en intraveineux, relayée per os avec dégression lente à laquelle s’associe fréquemment un immunosuppresseur tels que la cyclosporine, l’azathioprine, le méthotrexate ou le mycophenolate mofetil, permettant une épargne cortisonique, un meilleur contrôle de la maladie et un meilleur pronostic. La corticothérapie locale permet une guérison plus rapide de l’uvéite antérieure (8).
Les complications de la maladie comprennent une cataracte, un glaucome, une fibrose sous-rétinienne et des néo-vaisseaux choroïdiens pouvant compromettre le pronostic visuel. Par ailleurs, les complications infectieuses ne doivent pas être négligées comme en témoigne notre vignette clinique qui est le deuxième cas documenté dans la littérature illustrant une réactivation de tuberculose suite à un traitement immunosuppresseur pour maladie de VKH (9).
Conclusion
La maladie de VKH est rare, elle se traite par immunosuppression et une réactivation de tuberculose peut survenir dans le décours de la maladie. L’association des traitements immunosuppresseur et antituberculeux expose à de nombreux effets secondaires et interactions médicamenteuses pouvant être source d’accident ou d’échec thérapeutique. Comme illustré dans ce cas, le risque d’hépatotoxicité a motivé une réduction de la dose d’azathioprine pour débuter le traitement antituberculeux. La rifampicine, puissant inducteur du cytochrome p450, a mené à une réduction de l’effet thérapeutique de la methylprednisolone et à une récidive d’uvéite postérieure, habituellement très rare dans le décours de la maladie de Vogt Koyanagi Harada.
Recommandations pratiques
Ce cas clinique illustre l’importance d’évoquer un diagnostic alternatif infectieux en présence d’une uvéite postérieure d’évolution inhabituelle. La réactivation d’une tuberculose latente devra certainement être exclue en présence d’un traitement immunosuppresseur et/ou d’une corticothérapie à haute dose.
Une maladie de Vogt Koyanagi Harada, ou toute uvéite postérieure de longue durée, à laquelle s’ajoute une réactivation de tuberculose nécessite une prise en charge thérapeutique pluridisciplinaire, entre ophtalmologues et internistes, idéalement dans un centre de soin tertiaire.
Affiliations
1 Département d’ophtalmologie, Cliniques universitaires Saint-Luc
2 Département de médecine interne et maladies infectieuses, Cliniques Universitaires Saint-Luc
Correspondance
Dr Zineb Bouterfa
Clinique Saint-Pierre Ottignies
Ophtalmologie
Avenue Reine Fabiola 9
B-1340 Ottignies
zineb.bouterfa@cspo.be
Références
- Fardeau C, Tran TH, Gharbi B, Cassoux N, Bodaghi B, Lehoang P. Retinal fluorescence and indocyanine green angiography and optical coherence tomography in successive stages of Vogt-Koyanagi-Harada disease. Int opthalmol 2007; 27 : 163-72
- Arellanes-Garcia L, Hernandez-Barrios M, Fromow-Guerra J, Cervantes-Fanning P. Fluorescein fundus angiographyc findings in Vogt-Koyanagi-Harada syndrome. Int ophtalmol 2007; 27: 155-61
- McAllister WA, Thompson PJ, Al-Habet SM, Rogers HJ, Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J 1983; 286: 923–925.
- Chen J, Raymond K. Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob 2006; 5: 3.
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res 2016; 52: 84–111
- O’Keefe GA, Rao NA. Vogt-Koyanagi-Harada disease. Surv ophtalmol 2017 ; 62:1-25
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001; 131:647–652
- Concha-del Rio LE, Gomez L, Arellanes-Gacia L. Corticotherapy vs. Corticotherapy plus immunosuppressive therapy in acute Vogt-Koyanagi-Harada disease. Arch soc esp oftalmol 2018; 93:225-230
- Tian-wei Qian, Su-Qin Yu, Xun Xu. A challenging case of tuberculosis-associated uveitis after corticosteroid treatment for Vogt-Koyanagi-Harada disease. Int J ophtalmol 2018; 11: 1430–1432.