Introduction
Depuis une dizaine d’années, les termes « intersexualité » ou « ambiguïté sexuelle » ont été remplacés par « désordre du développement sexuel » ou « anomalie du développement sexuel », correspondant à tout aspect inhabituel des organes génitaux ou à une discordance entre le sexe chromosomique, gonadique et génital (1). Selon cette définition, la prévalence est de 5 pour 1000 naissances. L’anomalie la plus fréquemment observée est l’hypospadias chez 73% des garçons (1,2).
La découverte d’anomalies des organes génitaux externes lors des échographies anténatales sont des situations rares mais complexes. Elles nécessitent une approche multidisciplinaire spécialisée impliquant des obstétriciens, des néonatologues, des pédiatres, des endocrinologues, des chirurgiens, des urologues, des généticiens, des radiologues ainsi que des psychologues et des pédopsychiatres, afin d’éviter des erreurs et des retards dans le diagnostic. En effet, le suivi de patients atteints de désordres du développement sexuel (DSD) est un défi tant sur le plan médical que psycho-social (3).
Cas clinique
Madame C, âgée de 22 ans, est enceinte pour la première fois. La grossesse a été obtenue de manière spontanée. À 18 semaines de grossesse, étant donné la discordance entre les résultats du NIPT (sexe chromosomique masculin) et de l’échographie fœtale en faveur d’un phénotype plutôt féminin, une ponction de liquide amniotique a été réalisée à 20 semaines d’aménorrhée afin de confirmer le caryotype fœtal. De plus, un RCIU est diagnostiqué : le fœtus présente un poids fœtal estimé inférieur au percentile 1 et des fémurs courts.
Anamnèse familiale
Madame C est issue d’une fratrie de deux. Sa sœur a déjà trois enfants dont deux petites filles et un garçon. Le père de la patiente présenterait un diabète de type 1. Monsieur L, compagnon de Mme C est issu d’une fratrie de cinq. Sa sœur a un fils de 14 mois qui présenterait des crises d’épilepsie.
Antécédents médicaux du couple
Madame C présente un diabète de type 1 traité par une pompe à insuline. Le diabète est bien équilibré depuis un an. Elle travaille habituellement en tant qu’esthéticienne mais a stoppé son travail depuis le mois de novembre et n’est donc pas en contact avec des produits toxiques. Monsieur L est en bonne santé et ne présenterait pas d’antécédents médicochirurgicaux particuliers. Il n’y aurait pas de lien de parenté au sein du couple.
Examens complémentaires
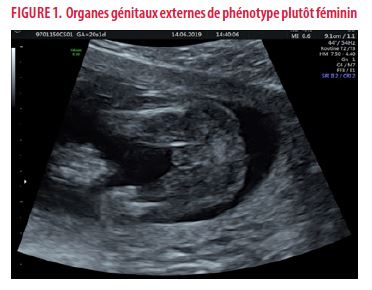
- À l’échographie réalisée à 20 semaines d’aménorrhée (SA), on note un RCIU, les biométries sont de 18-19SA. Le phénotype semble bien féminin. On ne visualise pas d’anomalie à l’examen morphologique du fœtus et des annexes fœtales [Figure 1].
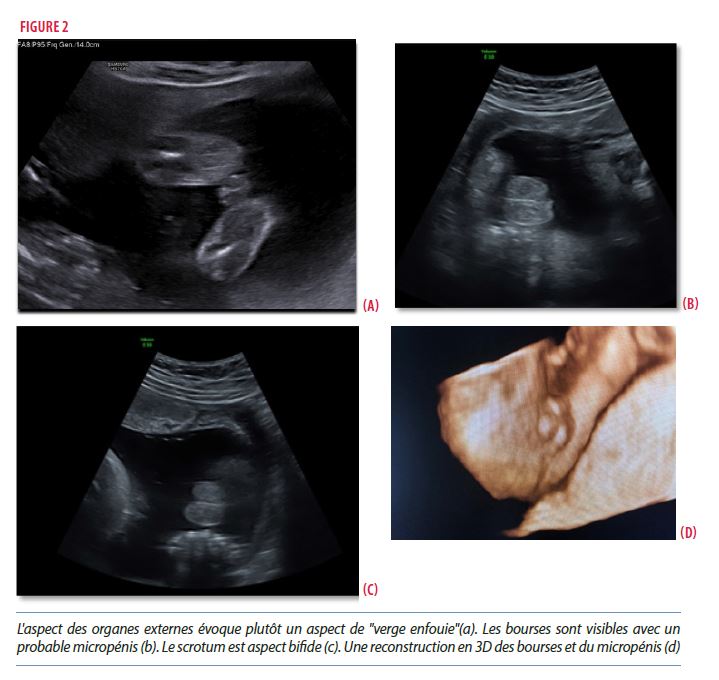
- À l’échographie morphologique du second trimestre (24SA), le RCIU est toujours présent avec une estimation du poids fœtal (EPF) inférieure au P1. Les 2 fémurs sont inférieurs au percentile 1 sans déformation. Au niveau morphologique, les omoplates sont d’aspect normal, il n’y a pas de rétrognathisme, le profil et le corps calleux sont visualisés, l’écart inter-orbitaire est normal. Il n’y a pas d’anomalie des extrémités mise en évidence. Il ne semble pas y avoir de structures interposées entre le rectum et la vessie, l’utérus n’est pas visualisé. L’aspect des organes externes évoque plutôt un aspect de «verge enfouie» que celui d’une hypertrophie clitoridienne, la miction n’a pas été observée en 1h30 pour éventuellement visualiser un hypospadias [Figure 2].
- À l’échographie morphologique du 3ème trimestre (32SA), la croissance fœtale se poursuit selon la courbe du P1 (EPF est de 1500g). Les os longs restent aussi sur leurs courbes <P3 [Figure 3]. Les bourses sont visibles avec un probable petit pénis. Il n’y a pas d’autres anomalies visualisées à l’examen morphologique du fœtus et des annexes fœtales.
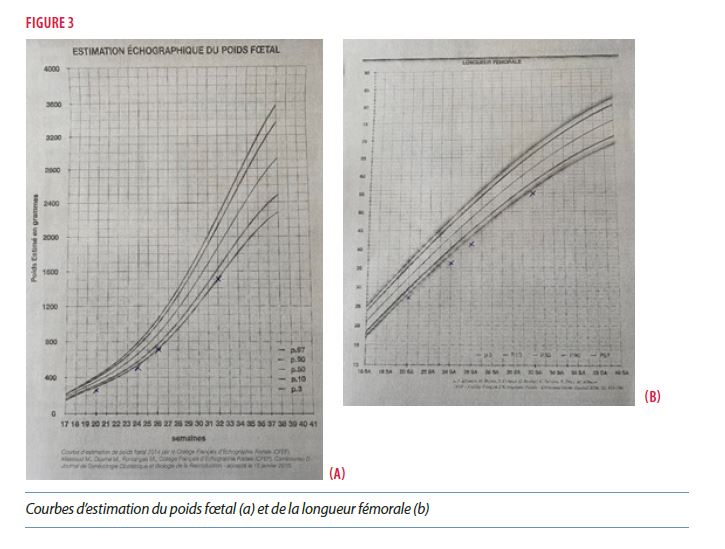
- Le scanner osseux 3D à 27SA montre des os longs courts de façon harmonieuse : la longueur fémorale est estimée à 41mm (VN 53 +/-3), les os de la jambe mesurent 35 mm (VN 45 +/-3). L’humérus mesure 40 mm (VN 49 +/-3). Les os de l’avant-bras mesurent 37 mm (VN43 +/-3). Il n’y a pas de malformation ou de déformation osseuse.
- Les prélèvements réalisés sur le liquide amniotique montrent un caryotype moléculaire normal : CGH-array : arr(1-22)x2,(X,Y)x1. Le dosage des stéroïdes révèle des valeurs normales avec un taux de testostérone satisfaisant et en accord avec un caryotype masculin. Au niveau génétique, le séquençage du gène SRY est également revenu normal. De même, le gène du récepteur aux androgènes ne montre pas de mutation. L’analyse des gènes SF-1 et WT1, facteurs impliqués dans la différenciation précoce de la gonade, n’a pas mis en évidence de mutation tout comme le panel DSD (disorder of sexual development) (Whole Exome Sequencing) dont l’analyse s’est avérée normale. Sur le plan syndromique, l’absence de mutation au sein du gène DHCR7 a permis d’exclure un syndrome de Smith-Lemli-Opitz et l’étude du gène SOX 9 (séquençage exons et MLPA) a permis d’écarter l’hypothèse d’une dysplasie acampomélique campomélique.
L’enfant est né à 36 semaines 2/7 à la suite d’une mise en travail spontanée. L’adaptation à la naissance a été tout à fait correcte en dehors d’un retard de résorption traité de façon transitoire par ventilation en pression positive continue (CPAP). À l’examen clinique, on note un hypospade péno-scrotal avec un scrotum bifide et un pénis mesuré à 1.7 cm. Les testicules sont palpés bilatéralement et de bon volume. L’échographie abdominale montre un aspect tout à fait normal. Le bilan hormonal réalisé à la naissance révèle une fonction testiculaire satisfaisante comme en témoignent les valeurs de testostérone (4.4 nmol) et de l’hormone antimüllérienne (AMH) (35 µg/l). On note des taux de LH et de FSH indosables en raison de l’imprégnation hormonale maternelle. Le delta 4 androsténedione et la DHEA sulfate sont augmentés, reflétant l’activité de la surrénale fœtale. La 17-OH-P est à 24.5 nmol/l (valeur normale vu la prématurité). La fonction thyroïdienne est également normale.
Un second bilan a été réalisé à la mini-puberté à 6 semaines de vie, lors d’une phase active de l’axe gonadotrope qui est probablement liée à l’interruption du feedback négatif des stéroïdes et peptides du placenta. Celui-ci a pu montrer une activation correcte de l’axe hypothalamo-hypophysaire puisque la LH est à 3.7 U/l avec une FSH à 1.3 U/l et une testostérone à 1.84 ng/ml. Le bilan biologique réalisé est donc satisfaisant avec un axe hypothalamo-hypophyso-gonadique qui semble actif et une fonction testiculaire qui est normale. En effet, l’AMH est à 129 µg/l ainsi que l’inhibine B qui est mesurée à 300.9 ng/l. De même, le contrôle de la 17-OH-P est à 0.84 ng/ml.
Un traitement par testostérone IM a été instauré à l’âge de 5 mois à raison de 3 injections de Sustanon® 25 mg 1x/mois avec un pénis mesuré à 3 cm après la réalisation des injections. La correction chirurgicale de l’hypospade sera prévue vers l’âge de 1 an.
Discussion
Le déterminisme sexuel dépend d’une cascade d’événements cellulaires et hormonaux, qui se déroule dans un ordre précis, mais encore mal défini, pour permettre le développement d’un appareil génital de morphotype masculin ou féminin.
La différenciation sexuelle se déroule en 4 étapes
- La première étape essentielle au développement normal des organes génitaux est la présence des chromosomes sexuels ou gonosomes. C’est l’établissement du sexe chromosomique et elle se constitue lors de la fécondation. Pour rappel, l’ovocyte maternel donne 23X et le spermatozoïde paternel donne soit 23X soit 23Y. Il en résulte un zygote homogamétique 46XX de sexe génétique féminin ou hétérogamétique 46XY de sexe génétique masculin. Il peut être différent du sexe gonadique ou phénotypique. C’est donc l’étape de fertilisation et de détermination du sexe chromosomique (2, 5-7).
- La seconde étape est le développement de la gonade primitive sous la dépendance de gène(s) autosomique(s) encore largement inconnus. Les gonades restent indifférenciées jusqu’à la sixième semaine. C’est la formation des organes communs aux deux sexes. Comme exemple de gènes candidats à l’agonadisme, nous avons le gène WT1 qui est responsable, s’il s’agit d’un variant pathogène, du syndrome de Denys-Drash (dysgénésie gonadique, sclérose mésangiale diffuse et tumeur de Wilms). Un autre candidat est le gène SF1 dont les mutations hétérozygotes sont responsables d’insuffisance surrénalienne et/ou de dysgénésie gonadique [2, 5-7, 8).
- La troisième étape correspond au déterminisme du sexe gonadique (différenciation des gonades). La gonade bipotentielle va devenir soit un testicule soit un ovaire. Le gène SRY situé sur l’Y est un des facteurs testiculaires indispensables au développement de la gonade en testicule. Mais évidemment, il est loin d’être le seul ; comme exemple, le gène SOX9 est responsable, lorsqu’il est muté, de dysplasie campomélique et de DSD. En l’absence du gène SRY, la gonade primitive évoluera en ovaire, de façon spontanée. Dans la différenciation de la gonade en ovaire, certains gènes sont impliqués (WNT4, RSPO1). (2, 5-7,9).
Les divers dysfonctionnements de ces étapes 2 et 3 aboutiront au développement de gonades ne correspondant pas au sexe chromosomique.
- La dernière et quatrième étape est la différenciation des organes génitaux externes et internes dépendante de la synthèse et de l’action des androgènes. Les gonades commencent à sécréter des hormones à partir de la huitième semaine. Spontanément, les canaux de Müller vont fusionner partiellement et se développer pour donner l’utérus, les trompes et le tiers supérieur du vagin et inversement, les canaux de Wolff vont involuer. En revanche, en présence de l’AMH, sécrétée par les cellules de Sertoli testiculaires, les canaux de Muller vont régresser. De plus, en présence de testostérone, sécrétée par les cellules de Leydig testiculaires, les canaux de Wolff donneront les épididymes, les canaux déférents et les vésicules séminales. De même, les organes génitaux externes ne se masculiniseront qu’en présence de la dihydrotestostérone (transformation de la testostérone par la 5 alpha-reductase) (2, 4-7).
La connaissance de la physiopathologie du développement des organes génitaux externes va permettre de proposer une classification des différents DSD rencontrés. La classification des DSD en fonction du caryotype est proposé sur ce tableau 1 (10).
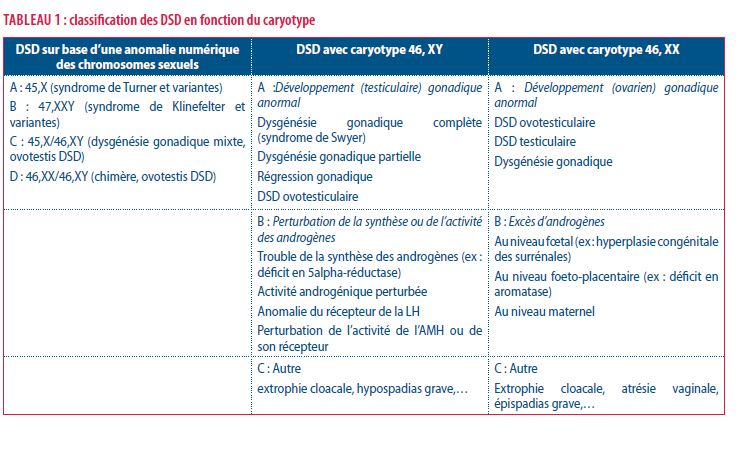
Néanmoins, la moitié des DSD restent, à l’heure actuelle, sans étiologie malgré un énorme travail de génotypage. Notons que pour un aspect inhabituel des OGE, 75% des cas sont porteurs d’un caryotype 46, XY, 10–15% sont porteurs d’un caryotype 46, XX (pour ces derniers, dans 95% des cas, la cause est une hyperplasie des surrénales) et 10% résultent d’une anomalie structurelle ou numérique des chromosomes sexuels (2).
Dans notre cas clinique, le désordre du développement sexuel a été diagnostiqué en anténatal, à 18 semaines de grossesse, et ce devant la discordance des résultats du NIPT (sexe chromosomique masculin) et de l’échographie fœtale en faveur d’un phénotype plutôt féminin. Depuis, la réalisation du NIPT qui est prescrit en routine, un plus grand nombre de DSD sont diagnostiqués en anténatal. En effet, via l’étude de l’ADN fœtal libre circulant dans le sang maternel, nous pouvons en plus du dépistage des trisomies 13, 18 et 21, connaître le sexe fœtal. Il en résulte un certain bénéfice, une prise en charge rapide à la naissance du nouveau-né avec une anomalie du développement sexuel au niveau étiologique et thérapeutique ainsi qu’une possibilité de permettre une déclaration du sexe plus rapidement (11). Cependant, lors de ce diagnostic anténatal, une question éthique a été soulevée par nos futurs parents : « c’est l’interruption médicale de grossesse ». Celle-ci peut être légitime face à l’angoisse rencontrée lors de l’annonce d’une anomalie chez son enfant, la difficulté de poser une étiologie bien définie, l’incertitude sur le devenir au long cours de l’enfant. La rencontre avec une équipe pluridisciplinaire aura aussi un rôle sur l’acceptation de la différence par les parents. Une communication la plus claire possible doit être favorisée afin de consolider la confiance du couple au sein de l’équipe médicale. Ce qui favorisera les prises de décision futures ainsi que la compliance thérapeutique qui en découlera.
Chez ce fœtus, différents examens complémentaires ont été réalisés durant la grossesse. Dans un premier temps, une ponction de liquide amniotique a montré un caryotype moléculaire normal masculin. Une anomalie au niveau de l’étape du sexe chromosomique a donc été exclue. Notons qu’une confirmation du caryotype à la naissance a été demandée et est aussi revenu normal, 46, XY (par caryotype standard pour rechercher une mosaïque ou un remaniement chromosomique équilibré non visible au caryotype moléculaire).
Les suivis échographiques fœtaux ont montré, en plus d’une mauvaise différenciation des organes génitaux externes masculins (aspect de «verge enfouie», bourses visibles avec probable micropénis, hypospade non exclu), un RCIU sévère (percentile 1) et des fémurs courts.
Différentes analyses génétiques ont été réalisées afin de rechercher un désordre du développement testiculaire, c’est-à-dire, la mise en place du sexe gonadique (étape 2 et 3) : gène SRY - gène SOX 9 - gène WT1 - gène SF1 et un Panel de gènes impliqués dans les DSD (Whole Exome Sequencing), aucune analyse n’a permis de mettre en évidence de mutation permettant d’expliquer le phénotype fœtal. Vu l’association du retard de croissance intra-utérin et de l’anomalie de développement génital, une mutation au niveau du gène DHCR7 (Syndrome de Smith-Lemli-Opitz) a été recherchée et s’est avérée négative. Les mutations du gène DHCR7 entraînent un déficit en 7-déhydrocholestérol réductase, enzyme qui convertit le 7-dehydrocholesterol (7DHC) en cholestérol.
Au niveau des examens hormonaux, le dosage des stéroïdes sur le liquide amniotique est normal avec un taux de testostérone satisfaisant et en accord avec le sexe chromosomique masculin. Le gène du récepteur aux androgènes n’a pas montré de mutation. De même, les bilans hormonaux réalisés à la naissance et à la mini-puberté montrent une fonction testiculaire correcte et un axe hypothalamo-hypophyso-gonadotrope fonctionnel (12).
Devant un cas clinique de DSD, les différentes étapes de la différenciation sexuelle permettent de comprendre l’origine de l’anomalie du sexe du patient dans une majorité des cas. Néanmoins, la compréhension des mécanismes de la physiopathologie du déterminisme du sexe reste incomplète et certaines situations cliniques demeurent non résolues. En effet, l’étiologie moléculaire peut aider à la prise en charge, mais, il existe une importante variabilité phénotypique pour une même mutation, probablement parfois en raison d’une affection multigénique. C’est pour cette raison qu’une étroite collaboration entre les centres de référence et de compétence « DSD » doit permettre d’harmoniser nos pratiques pour la prise en charge de ces pathologies rares. On ne retrouve un diagnostic moléculaire que dans 30-50% des cas de DSD à caryotype 46, XY (1-3, 5-9).
Pour ce cas clinique, l’étiologie d’un DSD idiopathique avec fonction testiculaire normale a été posé sur un RCIU sévère d’origine vasculaire en raison d’un diabète de type 1 chez sa mère. En effet, plusieurs études rétrospectives ont montré une plus grande fréquence d’hypospades chez les fœtus masculins de petit poids. Ces observations évoquent d’autres facteurs : vasculaires, les facteurs de croissance, une mauvaise synchronisation des différents facteurs de la différenciation sexuelle… mais aussi une origine placentaire. Différentes anomalies placentaires ont été retrouvées chez ce type de patient comme des infarcissements, des calcifications, des signes d’ischémie chronique. L’hypothèse de ce RCIU associé à un hypospade en raison d’une insuffisance placentaire serait une anomalie de la sécrétion de testostérone sous contrôle de l’hormone chorionique gonadotrope humaine (hCG) pendant les 14 premières semaines de gestation (6, 13-16).
Ces dernières années, de plus en plus d’études ont également émis l’hypothèse de l’implication possible de l’exposition aux perturbateurs endocriniens (PE) dans la survenue de désordre de la différenciation sexuelle, et en particulier dans la survenue des hypospades. L’exposition aux phtalates, PE à activité anti-androgénique, pourrait expliquer l’augmentation de l’incidence de ces malformations (17).
Par ailleurs, le diagnostic étiologique ne permet pas toujours de prédire l’identité sexuelle ou, communément, l’identité de genre. De nombreuses associations (Amnesty International, DSD-LIFE, Consensus Statement M Cools et al, 2018) ont fait pression pour différer les interventions chirurgicales ou traitements hormonaux invasifs et irréversibles chez des enfants présentant des variations des caractéristiques sexuelles jusqu’à ce qu’ils soient en mesure de prendre part de façon active à la décision et en connaissance de cause. Pour une réussite la plus optimale, il faut proposer à l’enfant, à l’adolescent ou à l’adulte, un traitement dont les possibilités de traitement chirurgical ou hormonal donnera le meilleur résultat au niveau psycho-social, psycho-sexuel et si possible au niveau de la fertilité, en tenant compte aussi du contexte culturel et de l’éventuelle imprégnation anténatale du cerveau par les androgènes (18).
Conclusion
Ce cas clinique anténatal de DSD nous rappelle la nécessité d’une prise en charge pluridisciplinaire et spécialisée, en raison de la rareté et de la complexité des cas rencontrés. L’étiologie retenue chez ce fœtus est un DSD idiopathique avec fonction testiculaire normale associé à un RCIU sévère d’origine vasculaire en raison d’un diabète de type 1 chez sa mère. À l’heure actuelle, la moitié des DSD reste sans étiologie. De nombreux mécanismes génétiques restent à découvrir. Le défi actuel de nos cliniciens, depuis l’arrivée du séquençage du génome, est un suivi au long cours de ces patients afin d’y intégrer le phénotypage clinique, biologique et anatomopathologique. Cela permettra dans un futur proche d’améliorer la classification des DSD et les propositions thérapeutiques.
Recommandations pratiques
La découverte d’anomalies des organes génitaux externes lors des échographies anténatales nécessite une approche multidisciplinaire en collaboration avec des centres de référence.
La connaissance des différentes étapes de la différenciation sexuelle permet de comprendre l’origine de l’anomalie du sexe dans une majorité des cas ; cependant, un diagnostic moléculaire n’est décrit que dans la moitié des cas.
Affiliations
(1) Chef de clinique adjoint, Service de Gynécologie-Obstétrique, CHU de Liège, site CHR Citadelle, Belgique.
(2) Chef de clinique adjoint, Service de génétique, CHU de Liège, Belgique.
(3) Chef de clinique, Service de pédiatrie, CHU de Liège, Belgique.
(4) Chef de service, Service de Gynécologie-Obstétrique, CHU de Liège, site CHR Citadelle, Belgique.
Correspondance
Dr. Cécile Habran
CHU de Liège, site CHR Citadelle
Chef de clinique adjoint
Service de Gynécologie-Obstétrique
Boulevard Emile de Laveleye 62
B-4020 Liège
Cecile.habran@chrcitadelle.be
Références
- Cartigny-Maciejewski M. Les désordres du développement sexuel. Dans: Letombe B, Catteau-Jonard S, Robin G, directeurs. Endocrinologie en gynécologie et obstétrique. Paris: Elsevier Masson; 2012. p. 13-25.
- Basé sur le cours de Mme Brachet C. Mécanismes de la différenciation sexuelle du 12 octobre 2018 dans le cadre d’un Certificat d’Université en Endocrinologie de la Reproduction Bruxelles, à l’Université Libre de Bruxelles.
- Phan-Hug F, Kraus C, Paoloni-Giacobino A, Fellmann F, Typaldou S-A, Ansermet F et al. Patients avec variations du développement sexuel : un exemple de prise en charge interdisciplinaire. Rev Med Suisse. 2016;12:1923-9. Disponible : https://www.revmed.ch/RMS/2016/RMS-N-538/Patients-avec-variation-du-developpement-sexuel-un-exemple-de-prise-en-charge-interdisciplinaire
- Hutson J, Grover S, O’Connell M, Pennell S. Malformation syndromes associated with disorders of sex development. Nat. Rev. Endocrinol. 2014;10:476-487. DOI:10.1038/nrendo.2014.83.
- Manouvrier-Hanu S. Faire un garçon ou une fille : mécanisme et gènes impliqués dans le déterminisme du sexe et la différenciation sexuelle. Dans: Letombe B, Catteau-Jonard S, Robin G, directeurs. Endocrinologie en gynécologie et obstétrique. Paris: Elsevier Masson; 2012. p. 3-11.
- Roucher F, Morel Y, Mallet D, Plotton I, Tardy V. Physiopathologie et classification des anomalies du développement génitosexuel. Rev. Méd. Périnat. 2015;7:137-146. DOI:10.1007/s12611-015-0336-6.
- Basé sur le cours de Mr Nef S. La détermination du sexe du 12 octobre 2018 dans le cadre d’un Certificat d’Université en Endocrinologie de la Reproduction Bruxelles, à l’Université Libre de Bruxelles.
- Fenichel P, Hieronimus S, Chevallier P, Cherikh F, Sultan C, Paris F. DSD 46,XY avec hypospadias et dysgénésie gonadique par mutation de SF1 (NR5A1) chez une jeune sportive de haut niveau dépistée par un taux élevé de testostérone circulante. Ann. Endocrinol. 2016;77(4):462. DOI:10.1016/j.ando.2016.07.624.
- Ono M, Harley V. Disorders of sex development: new genes, new concepts. Nat. Rev. Endocrinol. 2012;9(2):79-91. DOI:10.1038/nrendo.2012.235.
- Lee P, Houk C, Ahmed S, Hughes L. Consensus statement on management of intersex disorders. Arch. Dis. Child. 2006;91:1. DOI:10.1136/adc.2006.098319.
- Frade F, Peycelon M, Houang M, Hyon C, Siffroi J-P, Audry G. Anomalies du développement sexuel : les erreurs à ne pas faire à la maternité. Réal. Pédiatr [En ligne]. 2015. Disponible : http://www.realites-pediatriques.com/wp-content/uploads/sites/3/2016/04/RP_190_Dos_Frade.pdf.
- Becker M, Hesse V. Minipuberty: Why Does it Happen? Horm. Res. Paediatr. 2020;93:76-84. DOI:10.1159/000508329.
- Nordenvall A, Frisén L, Nordenström A, Lichtenstein P, Nordenskjöld A. Population based nationwide study of hypospadias in Sweden, 1973 to 2009 : incidence and risk factors. J Urol. 2014;191(3):783-9. DOI:10.1016/j.juro.2013.09.058.
- Cools M, Nordenström A, Robeva R, Hall J, Westerveld P, Flück C et al. Caring for individuals with a difference of sex development (DSD): a Consensus Statement. Nat. Rev. Endocrinol. 2018;14(7):415-429. DOI:10.1038/s41574-018-0010-8.
- Toufaily M, Roberts D, Westgate M-N, Hunt A-T, Holmes L. Hypospadias, Intrauterine Growth Restriction, and Abnormalities of the Placenta. Birth Defect Research. 2017;110(2):122-127. DOI:abs/10.1002/bdr2.1087.
- Fujimoto T, Suwa T, Kabe K, Adachi T, Nakabayashi M, Amamiya T. Placental insufficiency in early gestation is associated with hypospadias. J Pediatr Surg. 2008;43(2):358-61. DOI:10.1016/j.jpedsurg.2007.10.046.
- Raghavan R, Romano M, Karagas M, Penna F. Pharmacologic and Environmental Endocrine Disruptors in the Pathogenesis of Hypospadias: a Review. Environ Health Rep. 2018;5(4):499-511. DOI:10.1007/s40572-018-0214-z.
- Wilhelm D, Koopman P. The makings of maleness: towards an integrated view of male sexual development. Nature Reviews Genetics. 2006;7:620-631. DOI:10.1038/nrg1903.