INTRODUCTION
The anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA) is a rare congenital heart disease (incidence of 0,002%) (1,4). The physiopathology of ARCAPA is explained by retrograde blood flow in the RCA and the risk of myocardial ischemia. After birth, an inadequate collateralization can lead to death because myocardial perfusion is ensured by collateralization from the left coronary artery to the RCA, with a steal phenomenon to the pulmonary artery (1). Often asymptomatic in childhood (5), the diagnosis is easily missed and the anomaly can be incidentally diagnosed (6). However, ARCAPA may lead to myocardial ischemia and/or sudden cardiac death even in early childhood (5,7).
CASE REPORT
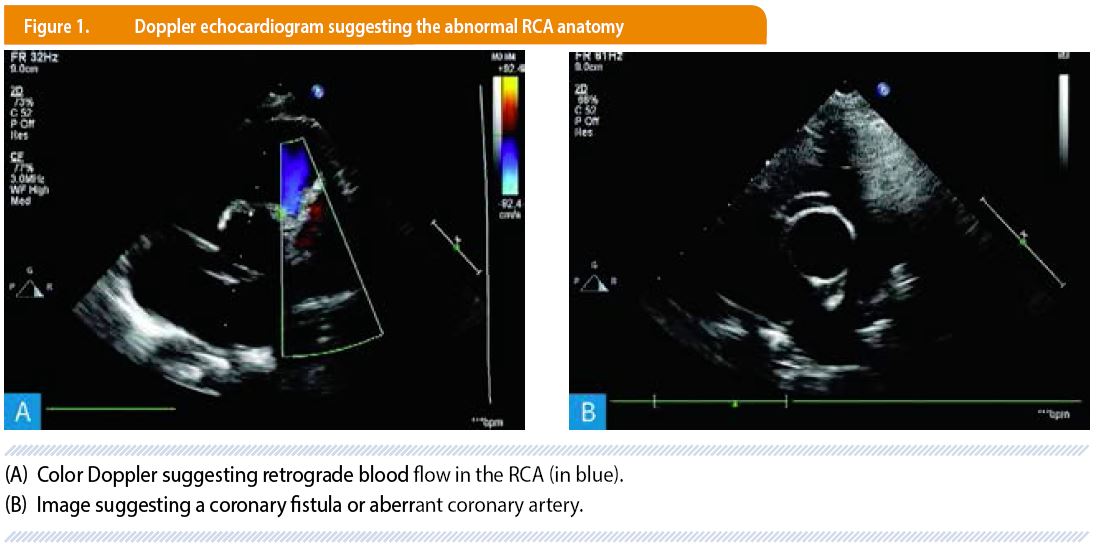
We report the case of a healthy 6 year-old boy with an isolated ARCAPA, revealed in the setting of a heart murmur. A first echocardiogram suggested the diagnosis of coronary fistula or aberrant coronary artery with a preserved systolic biventricular function. The patient’s clinical history, laboratory testings electrocardiogram and chest X-ray were otherwise unremarkable and the patient was referred to our center for further evaluation.
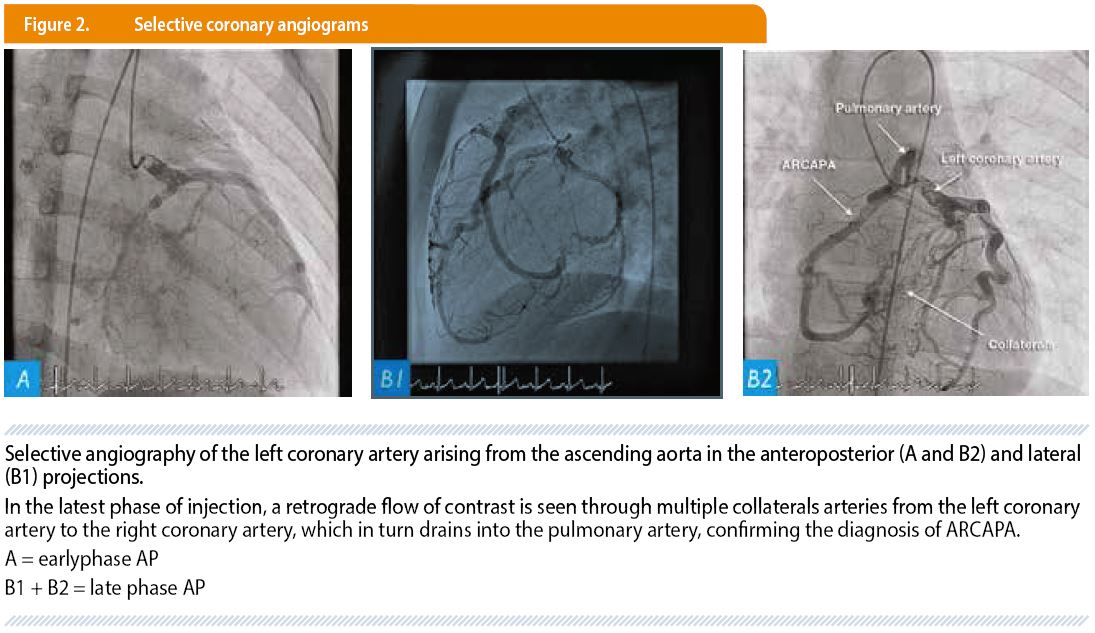
After a second color Doppler echocardiography (Figure 1), a cardiac catheterization was performed. Selective coronary angiograms showed a dilated left coronary system and a RCA arising from the main pulmonary artery and an extensive collateral circulation between the two coronary arteries with retrograde filling of the RCA from the left system (Figure 2). This confirmed the abnormal implantation of the RCA into the pulmonary artery.
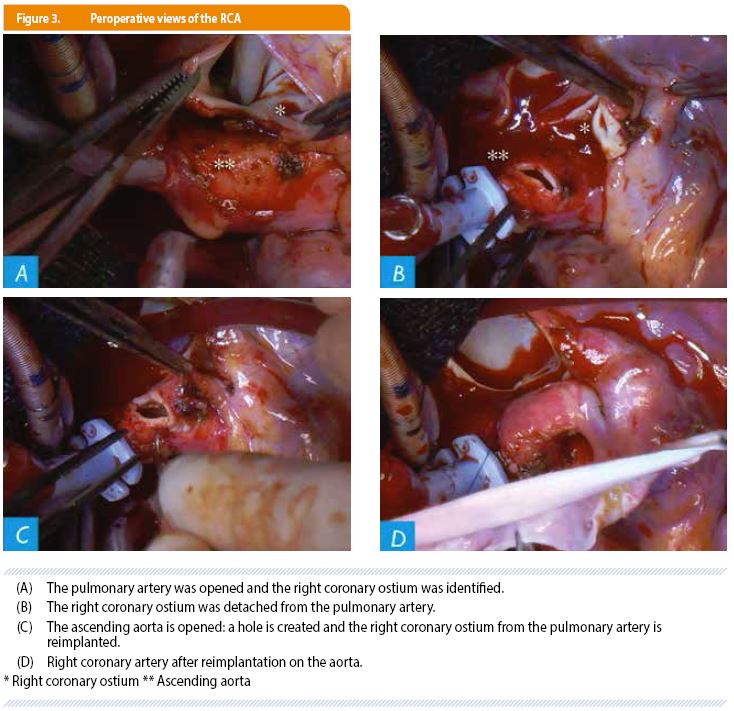
A re-implantation of the right coronary artery into the ascending aorta was performed in order to restore a normal dual coronary system (Figure 3). The procedure was uneventful and the patient was discharged on postoperative day 5.
An echocardiogram obtained at 1 year of follow-up demonstrated an unobstructed anterograde flow into the RCA.
DISCUSSION
Congenital anomalies of the coronary arteries are rare (< 1% of the pediatric population). Some of these abnormalities are benign but others are associated with a risk of myocardial ischemia or sudden death.
Among these congenital anomalies of the coronary arteries, there are:
- the anomalies of the origin of the coronary arteries;
- the anomalies of the course of the coronary arteries;
- the distal connection abnormalities mainly represented by coronary fistulas;
- the single coronary ostia (right or left) or the coronal duplication.
Isolated (70%) or associated with other heart defects (30%) like Tetralogy of Fallot or aorto-pulmonary window, ARCAPA is described as the abnormal origin of the RCA arising from the pulmonary artery instead of the right aortic sinus of Valsalva (1,7).
The physiopathology of ARCAPA is explained by the progressive development of a retrograde blood flow in the RCA when the pulmonary vascular resistance decreases after birth with a subsequent risk of myocardial ischemia (8).
In utero, the anomalous RCA arising from the pulmonary artery is perfused adequately because of the prenatal high pulmonary vascular resistance (1,3).
Shortly after birth, the pulmonary vascular resistance drops to significantly lower levels, and a significant collateralization between the right and left coronary arteries becomes mandatory to ensure sufficient myocardial perfusion (1,3). The ventricular ischemia in ARCAPA is less frequent than in patients with anomalous left coronary artery from the pulmonary artery (ALCAPA) because the right ventricle demands less oxygen than the left ventricle (8). ALCAPA is a similarly rare cardiac malformation characterized by an abnormal origin of the left coronary artery arising from the pulmonary artery, responsible for the development of heart failure, most often in infancy. Unlike ALCAPA (usually associated with signs of left ventricular ischemia and congestive heart failure in childhood (7)), patients with isolated ARCAPA are often asymptomatic in childhood and incidentally diagnosed. Some patients present heart murmur, chest pain, palpitations, dyspnea or rarely heart failure, arrhythmias, myocardial infarction or sudden death. Patients with associated heart anomalies are often diagnosed early (1,7).
The electrocardiogram is usually non-specific (7), unless it shows ischemic changes or left ventricular hypertrophy. The first-line investigation for the diagnosis of ARCAPA is echocardiography. Diagnostic features for ARCAPA include a dilated left coronary artery, intercoronary collateral arteries and a retrograde blood flow into the right coronary artery. The most common other diagnostic modalities are: angiography, cardio-CT and MRI (1,2,5). Angiography reveals the anomalous origin of the right coronary artery, as the contrast flows from the left system to the right system and subsequently drains into the main pulmonary artery. More recently, cardiac CT and MRI have provided three-dimensional artery coronary imaging with high diagnostic accuracy, with a spatial resolution still in favor of CT versus MRI (1,2).
A surgical correction is recommended even in asymptomatic patients based on a favorable risk-benefit ratio (1,3,4,5,8). It should be considered before the onset of ventricular dysfunction (4). In the literature, several techniques have been described with their advantages and disadvantages (8). The surgical alternatives for the reestablishment of dual coronary circulation are: a simple ligation or a percutaneous coil embolization of the RCA, a ligation and coronary bypass, the creation of an intrapulmonary baffle (Takeuchi operation), or a re-implantation of the right coronary artery into ascending aorta just above the normal implantation (4,5).
The aim of surgical correction is to reestablish dual coronary circulation (and eliminate the left-to-right shunt) to prevent the potential risk of myocardial ischemia from a coronary steal. The surgical re-implantation of the RCA to the ascending aorta in order to re-establish a physiologic double coronary artery system is the most commonly used in recent years, and has shown good long-term results (4,5).
CONCLUSION
ARCAPA, a rare congenital anomaly, is often asymptomatic in childhood and incidentally diagnosed. It’s associated with a risk of myocardial ischemia or sudden death. First-line investigation for the diagnosis is echocardiography. Other diagnostic modalities are: angiography, cardio-CT and MRI. Surgical correction is recommended even in asymptomatic patients based on a favorable risk-benefit ratio.
PRACTICAL RECOMMENDATIONS - TAKE HOME MESSAGE
• ARCAPA is often asymptomatic. Some patients present heart murmur, chest pain, palpitations, dyspnea or rarely heart failure, arrhythmias, myocardial infarction or sudden death.
• Risk of myocardial ischemia and sudden death.
• First-line investigation for the diagnosis of ARCAPA is echocardiography. Other diagnostic modalities are: angiography, cardio-CT and MRI.
• Surgical correction is recommended even in asymptomatic patients based on a favorable risk-benefit ratio.
AFFILIATIONS
1 Université catholique de Louvain, Cliniques universitaires Saint-Luc, Département de Cardiologie Pédiatrique, Bruxelles, Belgique.
2 Université catholique de Louvain, Cliniques universitaires Saint-Luc, Département de Chirurgie Cardiaque Pédiatrique, Bruxelles, Belgique.
CORRESPONDANCE
Dr. MORGANE SLAEDTS, MD
Cliniques universitaires Saint Luc
Pédiatrie
Avenue Hippocrate10, B-1200 Bruxelles, Belgium
morgane.slaedts@uclouvain.be
RÉFÉRENCES
- Kuba P-K, Sharma J, Sharma A. Successful surgical treatment of a septuagenarian with anomalous right coronary artery from the pulmonary artery with an eleven year follow-up: case report and review of literature. Sultan Qaboos University Med J. 2013; 13, 169–174. Open on PubMed
- Gilmour J, Hafka H, Ropchan G, Johri A-M. (2011). Anomalous Right Coronary Artery: A Multimodality Hunt for the Origin. Case Rep Cardiol. 2011;2011:286598. doi: 10.1155/2011/286598. Open on PubMed
- Williams I-A, Gersony W-M, Hellenbrand W-E. Anomalous right coronary artery arising from the pulmonary artery: a report of 7 cases and a review of the literature. Am Heart J. 2006; 152, 1004–1017. Open on PubMed
- Rajbanshi B-G, Burkhart H-M, Schaff H-V, et al. Surgical strategies for anomalous origin of coronary artery from pulmonary artery in adults. J Thoracic Cardiovasc Surg. 2014; 148, 220-224. Open on PubMed
- Chernogrivov A-E, Gornostaev A-A, Chernogrivov I-E, Toschev P-N, Suleimanov S-F, Talysheva O-N, et al. Anomalous origin of the right coronary artery from the pulmonary artery: surgical re-implantation into the aorta. Multimed Man Cardiothorac Surg. 2015 Sep 9;2015. pii: mmv024. doi: 10.1093/mmcts/mmv024. 2015 Open on PubMed
- De Bilderling G, Geurten C, Evrard J, Kanen F, Houtekie L, Carbonez K, et al. Quand l’insuffisance cardiaque se cache derrière la bronchiolite. Louvain Med 2017; 136 (4): 245-249.
- Kyu Seon K, Eun Young J, Jae Hyeon Y, et al. Anomalous right coronary artery from pulmonary artery discovered incidentally in an asymptomatic young infant. Korean J Pediatr. 2016; 59, S80-S83.
- Al-Dairy A, Rezaei Y, Pouraliakbar H, et al. Surgical repair for anomalous origin of the right coronary artery from the pulmonary artery. Korean Circul J. 2017; 47, 144-147. Open on PubMed